

RAGE is a key cellular target for Abeta-induced perturbation in Alzheimer's disease
- PMID: 22202057
- PMCID: PMC3687351
- DOI: 10.2741/s265
RAGE is a key cellular target for Abeta-induced perturbation in Alzheimer's disease
Abstract
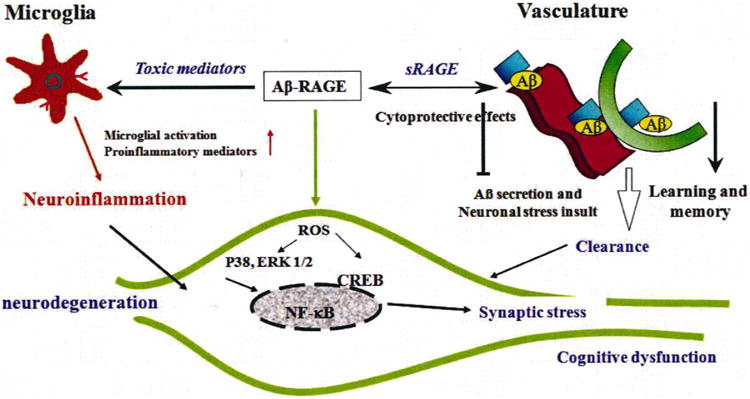
RAGE, a receptor for advanced glycation endproducts, is an immunoglobulin-like cell surface receptor that is often described as a pattern recognition receptor due to the structural heterogeneity of its ligand. RAGE is an important cellular cofactor for amyloid beta-peptide (Abeta)-mediated cellular perturbation relevant to the pathogenesis of Alzheimer's disease (AD). The interaction of RAGE with Abeta in neurons, microglia, and vascular cells accelerates and amplifies deleterious effects on neuronal and synaptic function. RAGE-dependent signaling contributes to Abeta-mediated amyloid pathology and cognitive dysfunction observed in the AD mouse model. Blockade of RAGE significantly attenuates neuronal and synaptic injury. In this review, we summarize the role of RAGE in the pathogenesis of AD, specifically in Abeta-induced cellular perturbation.
Figures
Similar articles
-
RAGE: a potential target for Abeta-mediated cellular perturbation in Alzheimer's disease.Curr Mol Med. 2007 Dec;7(8):735-42. doi: 10.2174/156652407783220741. Curr Mol Med. 2007. PMID: 18331231 Review.
-
RAGE-dependent signaling in microglia contributes to neuroinflammation, Abeta accumulation, and impaired learning/memory in a mouse model of Alzheimer's disease.FASEB J. 2010 Apr;24(4):1043-55. doi: 10.1096/fj.09-139634. Epub 2009 Nov 11. FASEB J. 2010. PMID: 19906677 Free PMC article.
-
Preventing activation of receptor for advanced glycation endproducts in Alzheimer's disease.Curr Drug Targets CNS Neurol Disord. 2005 Jun;4(3):249-66. doi: 10.2174/1568007054038210. Curr Drug Targets CNS Neurol Disord. 2005. PMID: 15975028 Review.
-
RAGE and Alzheimer's disease: a progression factor for amyloid-beta-induced cellular perturbation?J Alzheimers Dis. 2009;16(4):833-43. doi: 10.3233/JAD-2009-1030. J Alzheimers Dis. 2009. PMID: 19387116 Free PMC article. Review.
-
Involvement of microglial receptor for advanced glycation endproducts (RAGE) in Alzheimer's disease: identification of a cellular activation mechanism.Exp Neurol. 2001 Sep;171(1):29-45. doi: 10.1006/exnr.2001.7732. Exp Neurol. 2001. PMID: 11520119
Cited by
-
Plasma Amyloid-Beta Levels in a Pre-Symptomatic Dutch-Type Hereditary Cerebral Amyloid Angiopathy Pedigree: A Cross-Sectional and Longitudinal Investigation.Int J Mol Sci. 2021 Mar 13;22(6):2931. doi: 10.3390/ijms22062931. Int J Mol Sci. 2021. PMID: 33805778 Free PMC article.
-
Aβ42 oligomer-specific antibody ALZ-201 reduces the neurotoxicity of Alzheimer's disease brain extracts.Alzheimers Res Ther. 2022 Dec 29;14(1):196. doi: 10.1186/s13195-022-01141-1. Alzheimers Res Ther. 2022. PMID: 36578089 Free PMC article.
-
Pathophysiology of RAGE in inflammatory diseases.Front Immunol. 2022 Jul 29;13:931473. doi: 10.3389/fimmu.2022.931473. eCollection 2022. Front Immunol. 2022. PMID: 35967420 Free PMC article. Review.
-
Interaction between therapeutic interventions for Alzheimer's disease and physiological Aβ clearance mechanisms.Front Aging Neurosci. 2015 May 5;7:64. doi: 10.3389/fnagi.2015.00064. eCollection 2015. Front Aging Neurosci. 2015. PMID: 25999850 Free PMC article. Review.
-
Role of RAGE in Alzheimer's Disease.Cell Mol Neurobiol. 2016 May;36(4):483-95. doi: 10.1007/s10571-015-0233-3. Epub 2015 Jul 15. Cell Mol Neurobiol. 2016. PMID: 26175217 Free PMC article. Review.
References
-
- Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15:1437–1449. - PubMed
-
- Hansson Petersen CA, Alikhani N, Behbahani H, Wiehager B, Pavlov PF, Alafuzoff I, Leinonen V, Ito A, Winblad B, Glaser E, Ankarcrona M. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci U S A. 2008;105:13145–13150. - PMC - PubMed
-
- Eckert A, Hauptmann S, Scherping I, Meinhardt J, Rhein V, Drose S, Brandt U, Fandrich M, Muller WE, Gotz J. Oligomeric and fibrillar species of beta-amyloid (A beta 42) both impair mitochondrial function in P301L tau transgenic mice. Journal of molecular medicine (Berlin, Germany) 2008;86:1255–1267. - PubMed
-
- Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat Med. 2008;14:1097–1105. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
