

RB1 mutations and second primary malignancies after hereditary retinoblastoma
- PMID: 22205104
- PMCID: PMC3365233
- DOI: 10.1007/s10689-011-9505-3
RB1 mutations and second primary malignancies after hereditary retinoblastoma
Abstract
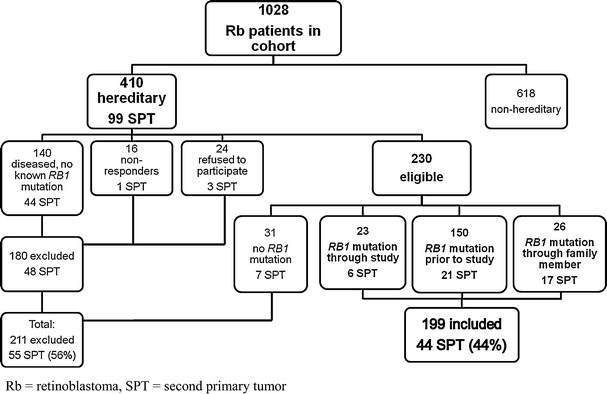
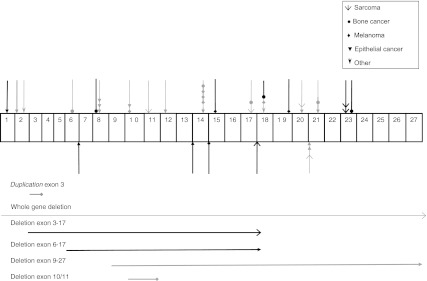
Survivors of hereditary retinoblastoma have a high risk of second primary malignancies, but it has not been investigated whether specific RB1 germline mutations are associated with greater risk of second primary malignancies in a large cohort. We conducted a retrospective cohort study of 199 survivors of hereditary retinoblastoma with a documented RB1 germline mutation diagnosed between 1905 and 2005. In total, 44 hereditary retinoblastoma survivors developed a second primary malignancy after a median follow-up of 30.2 years (range 1.33-76.0). A significantly increased risk of second primary malignancy was observed among carriers of one of the 11 recurrent CGA>TGA nonsense RB1 mutations (hazard ratio (HR) = 3.53; [95% confidence interval (CI) = 1.82-6.84]; P = .000), and there was a significantly lower risk for subjects with a low penetrance mutation (HR = .19; [95% CI = .05-.81]; P = .025). Our findings suggest a genotype-phenotype correlation for second primary cancers of retinoblastoma survivors and may impact on long-term surveillance protocols of patients with hereditary retinoblastoma, if confirmed by future studies.
Figures
Similar articles
-
Risk of second malignancies in survivors of retinoblastoma: more than 40 years of follow-up.J Natl Cancer Inst. 2008 Dec 17;100(24):1771-9. doi: 10.1093/jnci/djn394. Epub 2008 Dec 9. J Natl Cancer Inst. 2008. PMID: 19066271
-
Variation of second cancer risk by family history of retinoblastoma among long-term survivors.J Clin Oncol. 2012 Mar 20;30(9):950-7. doi: 10.1200/JCO.2011.37.0239. Epub 2012 Feb 21. J Clin Oncol. 2012. PMID: 22355046 Free PMC article.
-
The impact of RB1 genotype on incidence of second tumours in heritable retinoblastoma.Eur J Cancer. 2020 Jul;133:47-55. doi: 10.1016/j.ejca.2020.04.005. Epub 2020 May 17. Eur J Cancer. 2020. PMID: 32434110
-
Retinoblastoma: revisiting the model prototype of inherited cancer.Am J Med Genet C Semin Med Genet. 2004 Aug 15;129C(1):23-8. doi: 10.1002/ajmg.c.30024. Am J Med Genet C Semin Med Genet. 2004. PMID: 15264269 Review.
-
Retinoblastoma.Nat Rev Dis Primers. 2015 Aug 27;1:15021. doi: 10.1038/nrdp.2015.21. Nat Rev Dis Primers. 2015. PMID: 27189421 Free PMC article. Review.
Cited by
-
Subsequent Malignant Neoplasms in Retinoblastoma Survivors.Cancers (Basel). 2021 Mar 10;13(6):1200. doi: 10.3390/cancers13061200. Cancers (Basel). 2021. PMID: 33801943 Free PMC article. Review.
-
Retinoblastoma and Neuroblastoma Predisposition and Surveillance.Clin Cancer Res. 2017 Jul 1;23(13):e98-e106. doi: 10.1158/1078-0432.CCR-17-0652. Clin Cancer Res. 2017. PMID: 28674118 Free PMC article. Review.
-
Germline variants associated with leukocyte genes predict tumor recurrence in breast cancer patients.NPJ Precis Oncol. 2019 Nov 1;3:28. doi: 10.1038/s41698-019-0100-7. eCollection 2019. NPJ Precis Oncol. 2019. PMID: 31701019 Free PMC article.
-
Osteosarcoma without prior retinoblastoma related to RB1 low-penetrance germline pathogenic variants: A novel type of RB1-related hereditary predisposition syndrome?Mol Genet Genomic Med. 2019 Dec;7(12):e913. doi: 10.1002/mgg3.913. Epub 2019 Sep 30. Mol Genet Genomic Med. 2019. PMID: 31568710 Free PMC article.
-
Recommendations for Long-Term Follow-up of Adults with Heritable Retinoblastoma.Ophthalmology. 2020 Nov;127(11):1549-1557. doi: 10.1016/j.ophtha.2020.05.024. Epub 2020 May 15. Ophthalmology. 2020. PMID: 32422154 Free PMC article. Review.
References
-
- Mahoney MC, Burnett WS, Majerovics A, Tanenbaum H. The epidemiology of ophthalmic malignancies in New York State. Ophthalmology. 1990;97:1143–1147. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
