

Co-circulation of diverse paramyxoviruses in an urban African fruit bat population
- PMID: 22205718
- PMCID: PMC3542712
- DOI: 10.1099/vir.0.039339-0
Co-circulation of diverse paramyxoviruses in an urban African fruit bat population
Abstract
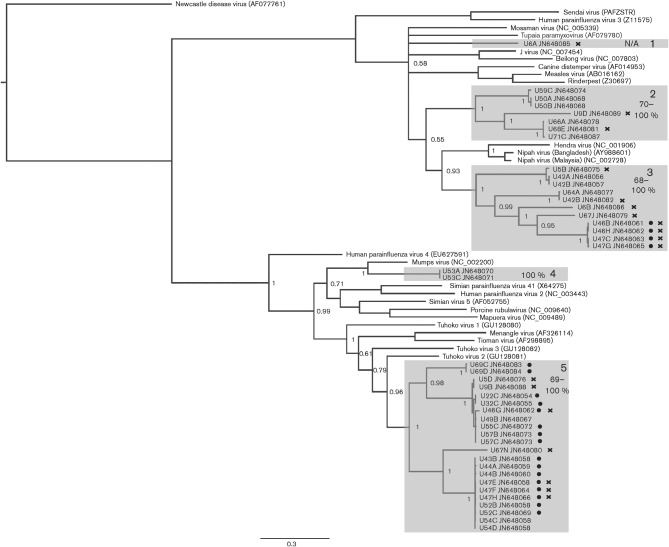
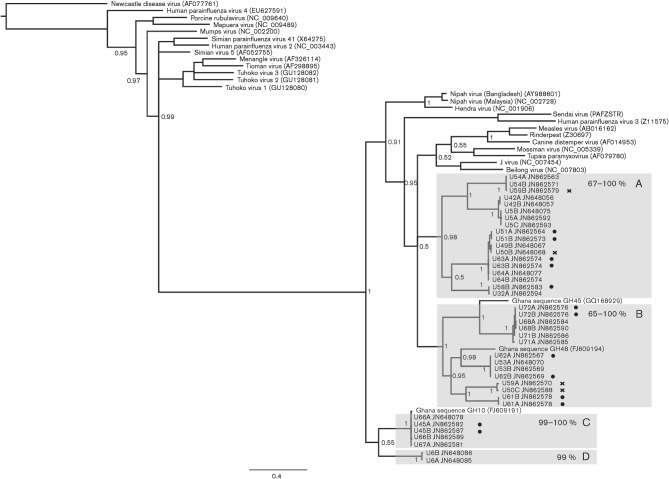
Bats constitute a reservoir of zoonotic infections and some bat paramyxoviruses are capable of cross-species transmission, often with fatal consequences. Determining the level of viral diversity in reservoir populations is fundamental to understanding and predicting viral emergence. This is particularly relevant for RNA viruses where the adaptive mutations required for cross-species transmission can be present in the reservoir host. We report the use of non-invasively collected, pooled, neat urine samples as a robust sample type for investigating paramyxoviruses in bat populations. Using consensus PCR assays we have detected a high incidence and genetic diversity of novel paramyxoviruses in an urban fruit bat population over a short period of time. This may suggest a similarly unique relationship between bats and the members of the family Paramyxoviridae as proposed for some other viral families. Additionally, the high rate of bat-human contact at the study site calls for the zoonotic potential of the detected viruses to be investigated further.
Figures
References
Publication types
MeSH terms
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources