

The human cytomegalovirus protein TRS1 inhibits autophagy via its interaction with Beclin 1
- PMID: 22205736
- PMCID: PMC3302257
- DOI: 10.1128/JVI.05746-11
The human cytomegalovirus protein TRS1 inhibits autophagy via its interaction with Beclin 1
Erratum in
-
Correction for Chaumorcel et al., "The Human Cytomegalovirus Protein TRS1 Inhibits Autophagy via Its Interaction with Beclin 1".J Virol. 2018 Apr 13;92(9):e00252-18. doi: 10.1128/JVI.00252-18. Print 2018 May 1. J Virol. 2018. PMID: 29654222 Free PMC article. No abstract available.
Abstract
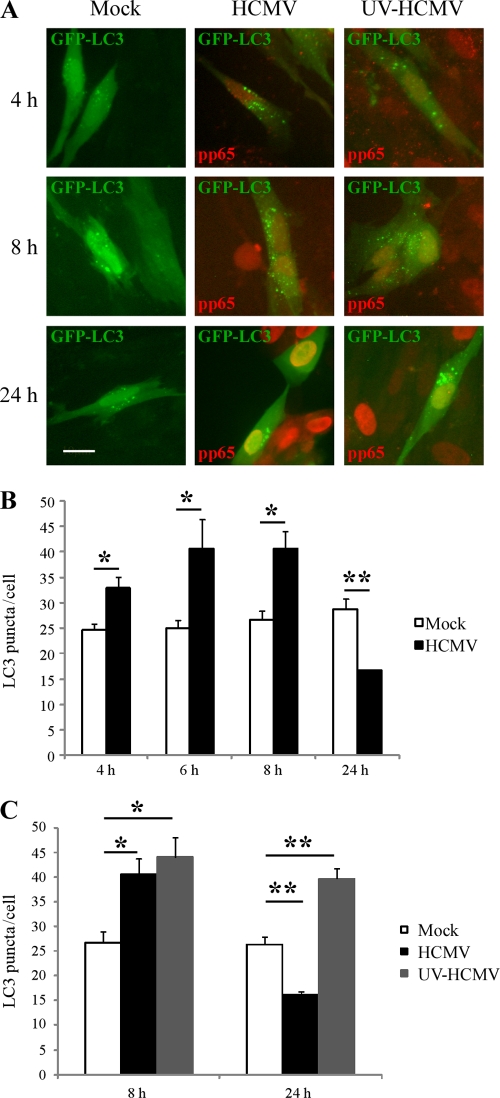
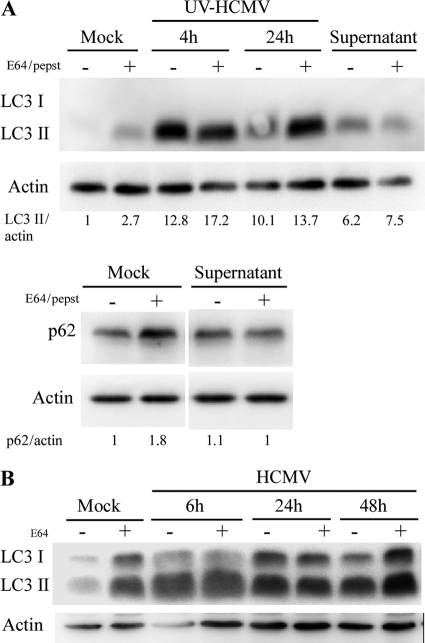
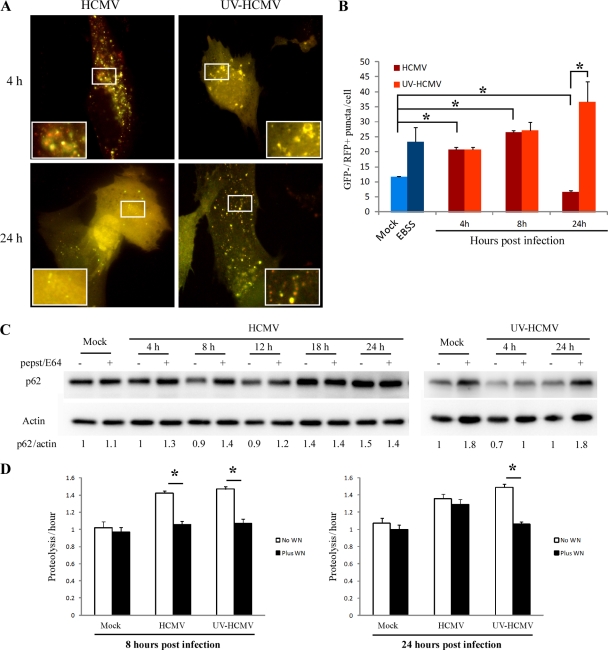
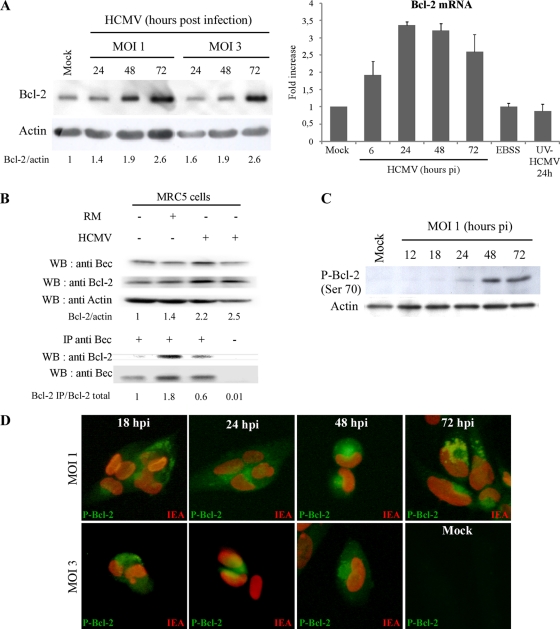
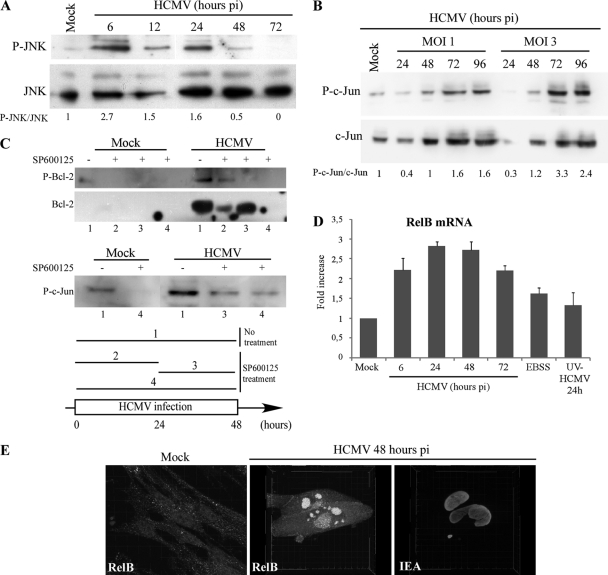
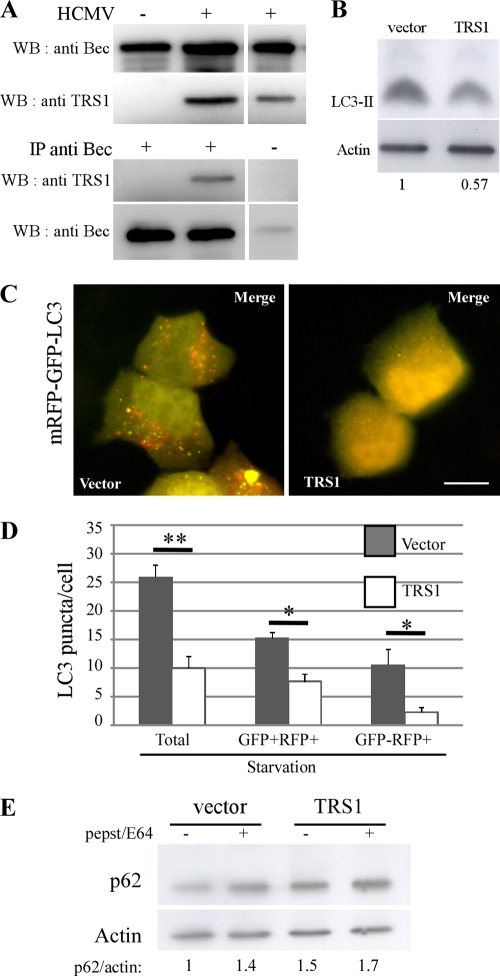
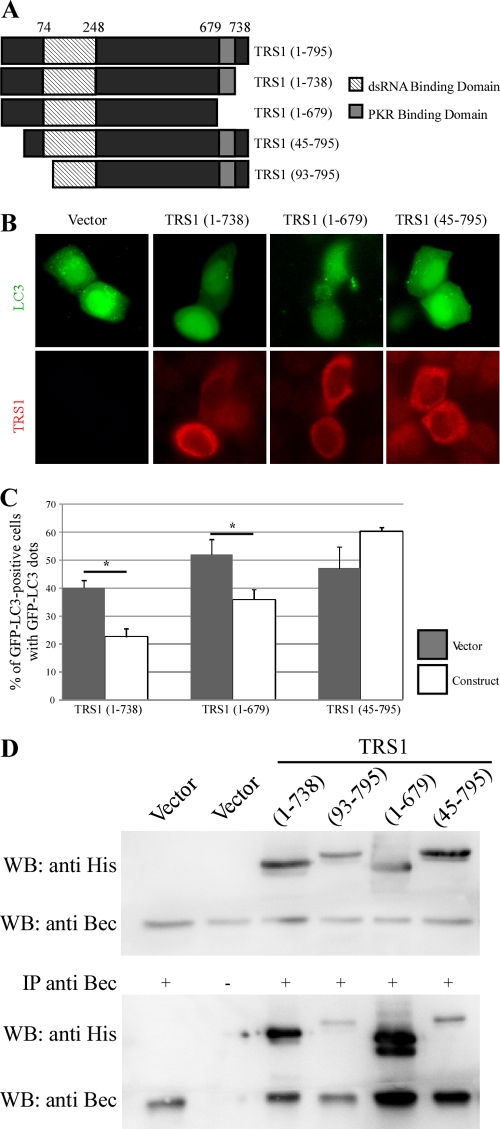
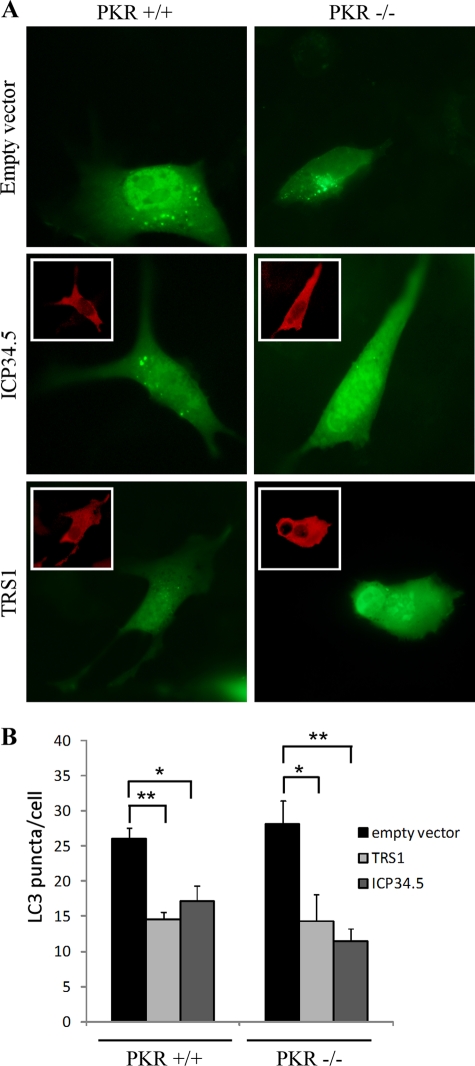
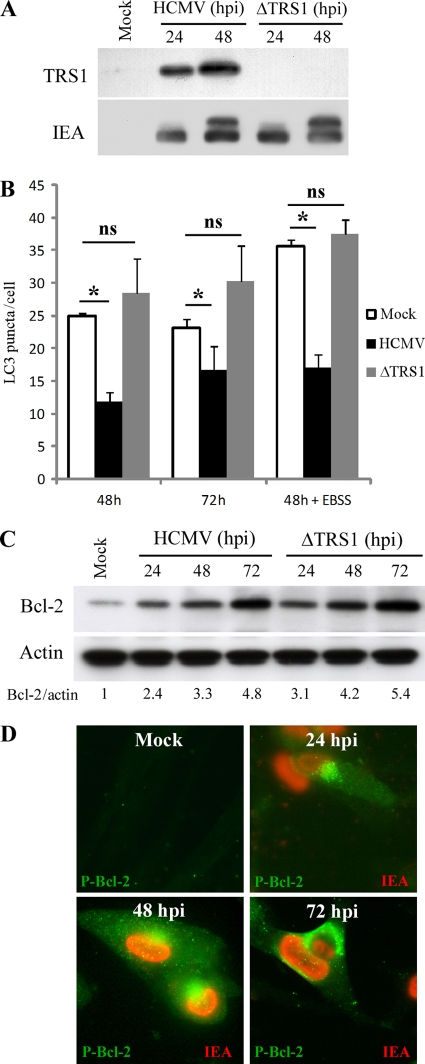
Human cytomegalovirus modulates macroautophagy in two opposite directions. First, HCMV stimulates autophagy during the early stages of infection, as evident by an increase in the number of autophagosomes and a rise in the autophagic flux. This stimulation occurs independently of de novo viral protein synthesis since UV-inactivated HCMV recapitulates the stimulatory effect on macroautophagy. At later time points of infection, HCMV blocks autophagy (M. Chaumorcel, S. Souquere, G. Pierron, P. Codogno, and A. Esclatine, Autophagy 4:1-8, 2008) by a mechanism that requires de novo viral protein expression. Exploration of the mechanisms used by HCMV to block autophagy unveiled a robust increase of the cellular form of Bcl-2 expression. Although this protein has an anti-autophagy effect via its interaction with Beclin 1, it is not responsible for the inhibition induced by HCMV, probably because of its phosphorylation by c-Jun N-terminal kinase. Here we showed that the HCMV TRS1 protein blocks autophagosome biogenesis and that a TRS1 deletion mutant is defective in autophagy inhibition. TRS1 has previously been shown to neutralize the PKR antiviral effector molecule. Although phosphorylation of eIF2α by PKR has been described as a stimulatory signal to induce autophagy, the PKR-binding domain of TRS1 is dispensable to its inhibitory effect. Our results show that TRS1 interacts with Beclin 1 to inhibit autophagy. We mapped the interaction with Beclin 1 to the N-terminal region of TRS1, and we demonstrated that the Beclin 1-binding domain of TRS1 is essential to inhibit autophagy.
Figures
References
-
- Bauvy C, Meijer AJ, Codogno P. 2009. Assaying of autophagic protein degradation. Methods Enzymol. 452:47–61 - PubMed
-
- Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelarova H, Meijer AJ. 1997. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur. J. Biochem. 243:240–246 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
