

Contribution of intra- and interhost dynamics to norovirus evolution
- PMID: 22205753
- PMCID: PMC3302298
- DOI: 10.1128/JVI.06712-11
Contribution of intra- and interhost dynamics to norovirus evolution
Abstract
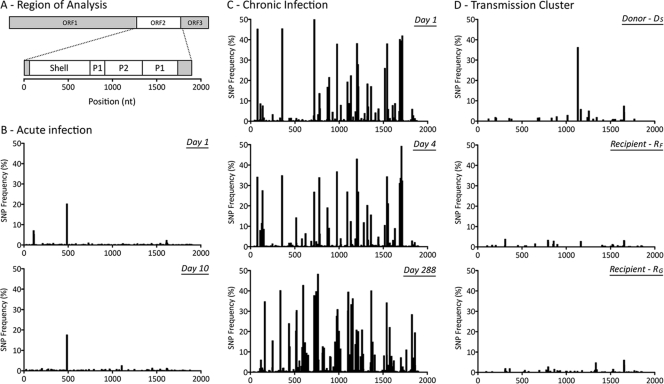
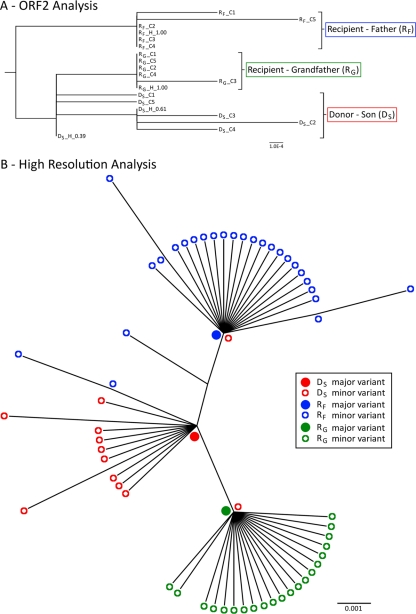
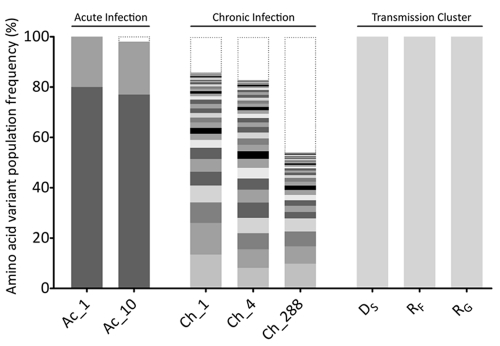
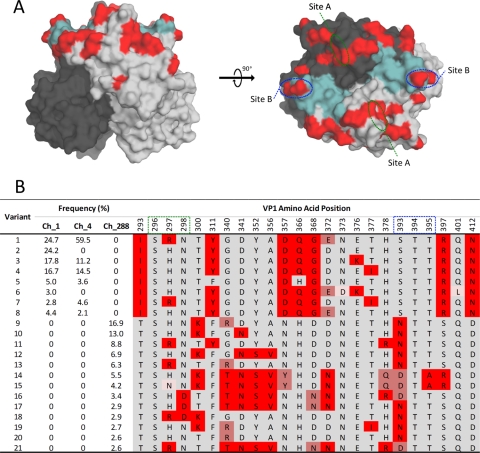
Norovirus (NoV) is an emerging RNA virus that has been associated with global epidemics of gastroenteritis. Each global epidemic arises with the emergence of novel antigenic variants. While the majority of NoV infections are mild and self-limiting, in the young, elderly, and immunocompromised, severe and prolonged illness can result. As yet, there is no vaccine or therapeutic treatment to prevent or control infection. In order to design effective control strategies, it is important to understand the mechanisms and source of the new antigenic variants. In this study, we used next-generation sequencing (NGS) technology to investigate genetic diversification in three contexts: the impact of a NoV transmission event on viral diversity and the contribution to diversity of intrahost evolution over both a short period of time (10 days), in accordance with a typical acute NoV infection, and a prolonged period of time (288 days), as observed for NoV chronic infections of immunocompromised individuals. Investigations of the transmission event revealed that minor variants at frequencies as low as 0.01% were successfully transmitted, indicating that transmission is an important source of diversity at the interhost level of NoV evolution. Our results also suggest that chronically infected immunocompromised subjects represent a potential reservoir for the emergence of new viral variants. In contrast, in a typical acute NoV infection, the viral population was highly homogenous and relatively stable. These results indicate that the evolution of NoV occurs through multiple mechanisms.
Figures

References
-
- Arnold K, Bordoli L, Kopp J, Schwede T. 2006. The SWISS-MODEL workspace: a Web-based environment for protein structure homology modelling. Bioinformatics 22:195–201 - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials
