

Reducing system noise in copy number data using principal components of self-self hybridizations
- PMID: 22207624
- PMCID: PMC3271883
- DOI: 10.1073/pnas.1106233109
Reducing system noise in copy number data using principal components of self-self hybridizations
Abstract
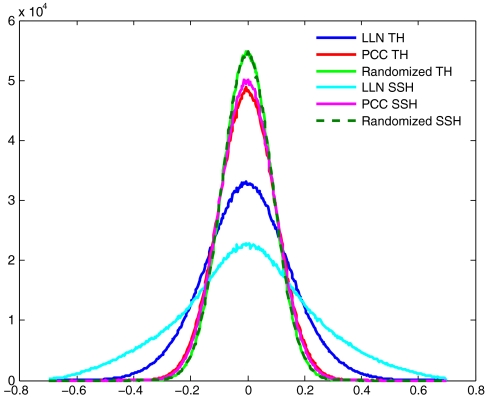
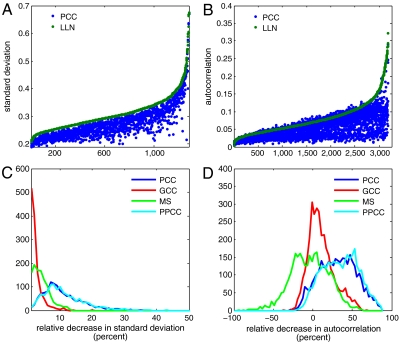
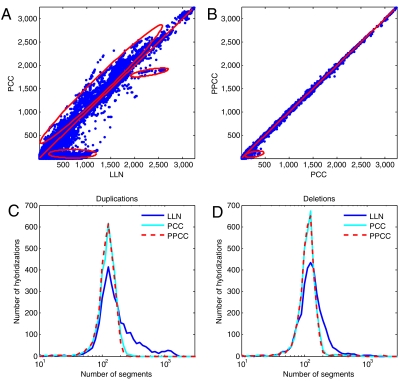
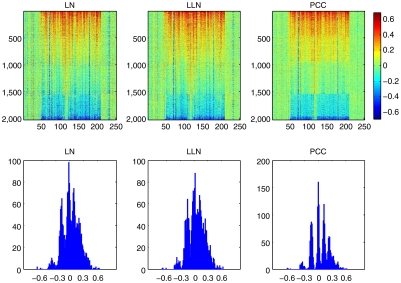
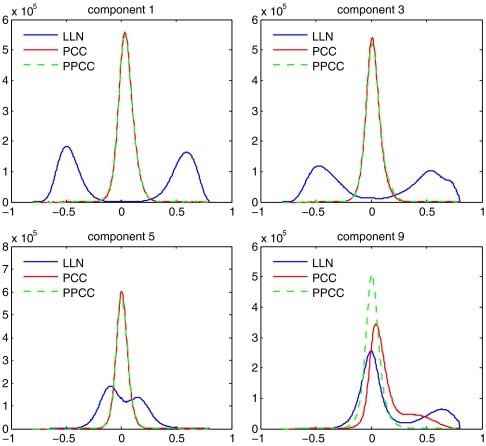
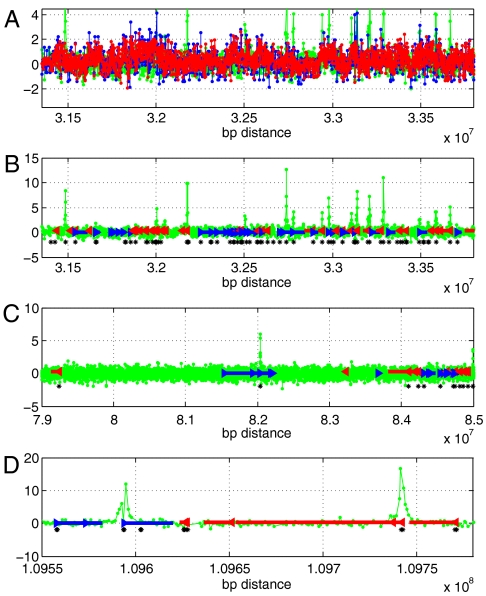
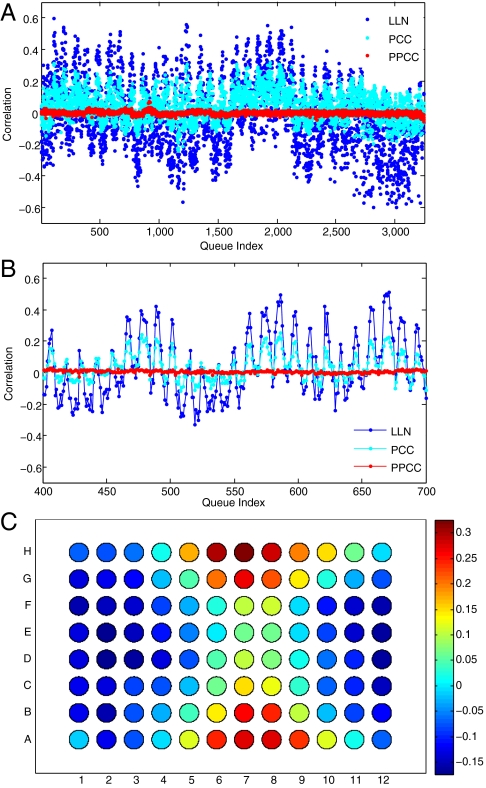
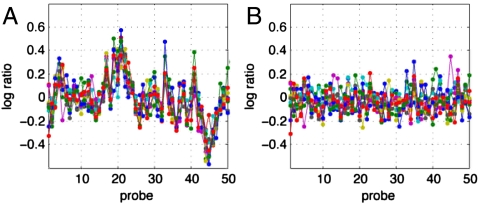
Genomic copy number variation underlies genetic disorders such as autism, schizophrenia, and congenital heart disease. Copy number variations are commonly detected by array based comparative genomic hybridization of sample to reference DNAs, but probe and operational variables combine to create correlated system noise that degrades detection of genetic events. To correct for this we have explored hybridizations in which no genetic signal is expected, namely "self-self" hybridizations (SSH) comparing DNAs from the same genome. We show that SSH trap a variety of correlated system noise present also in sample-reference (test) data. Through singular value decomposition of SSH, we are able to determine the principal components (PCs) of this noise. The PCs themselves offer deep insights into the sources of noise, and facilitate detection of artifacts. We present evidence that linear and piecewise linear correction of test data with the PCs does not introduce detectable spurious signal, yet improves signal-to-noise metrics, reduces false positives, and facilitates copy number determination.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Iafrate AJ, et al. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–951. - PubMed
-
- Sebat J, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. - PubMed
-
- Nei M, Niimura Y, Nozawa M. The evolution of animal chemosensory receptor gene repertoires: Roles of chance and necessity. Nat Rev Genet. 2008;9:951–963. - PubMed
-
- Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annu Rev Med. 2010;61:437–455. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
