

Acute exposure to low glucose rapidly induces endothelial dysfunction and mitochondrial oxidative stress: role for AMP kinase
- PMID: 22207730
- PMCID: PMC3319449
- DOI: 10.1161/ATVBAHA.111.227389
Acute exposure to low glucose rapidly induces endothelial dysfunction and mitochondrial oxidative stress: role for AMP kinase
Abstract
Objective: Hypoglycemia is associated with increased mortality. The reasons for this remain unclear, and the effects of low glucose exposure on vascular endothelial function remain largely unknown. We endeavored to determine the effects of low glucose on endothelial cells and intact human arterioles.
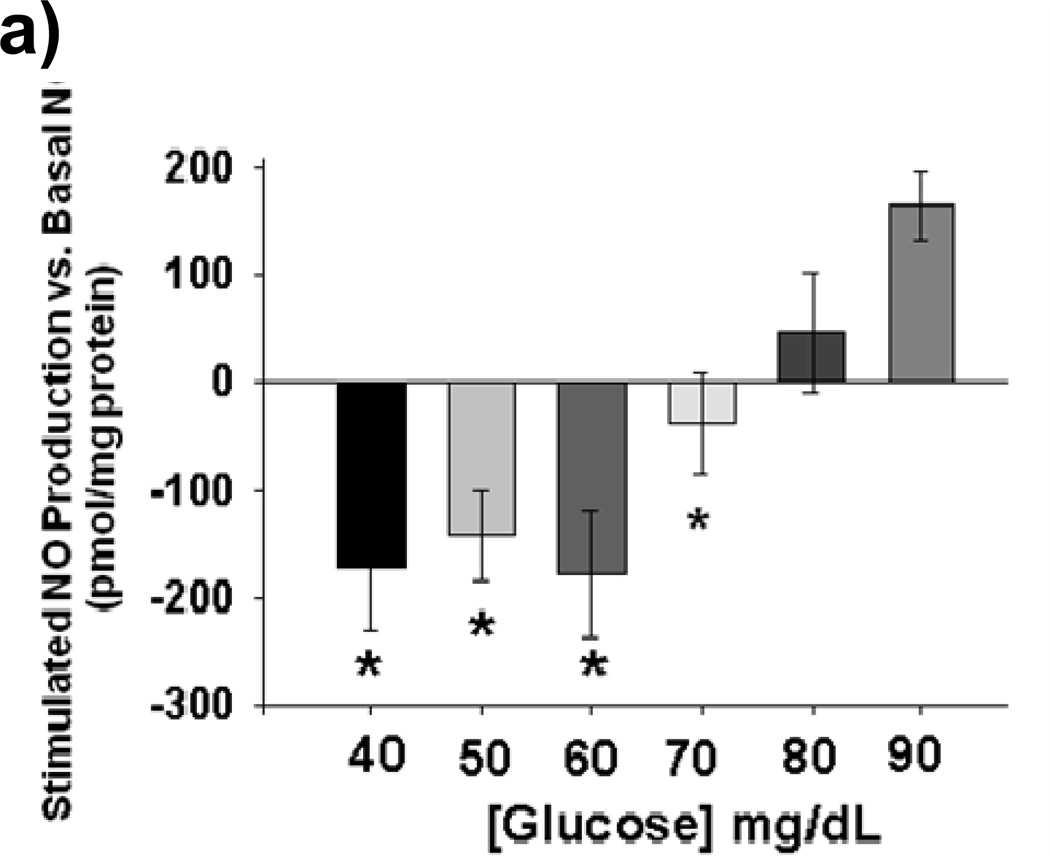
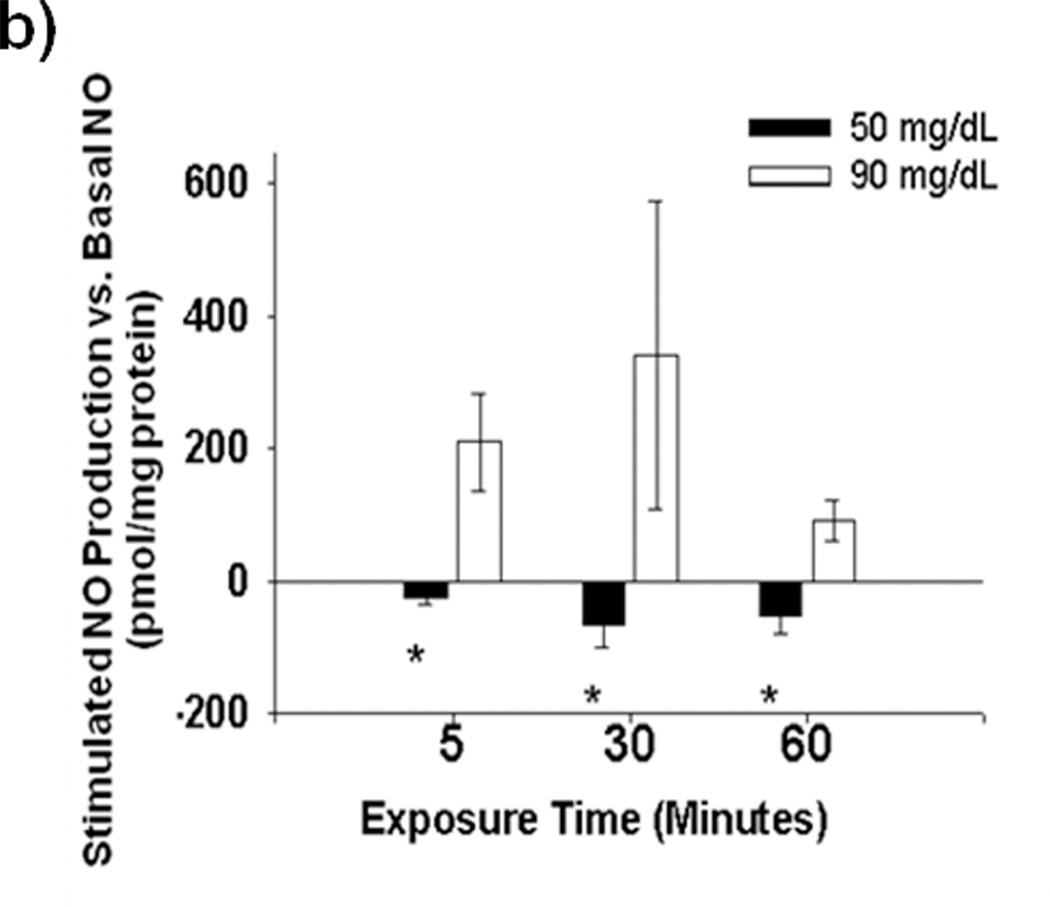
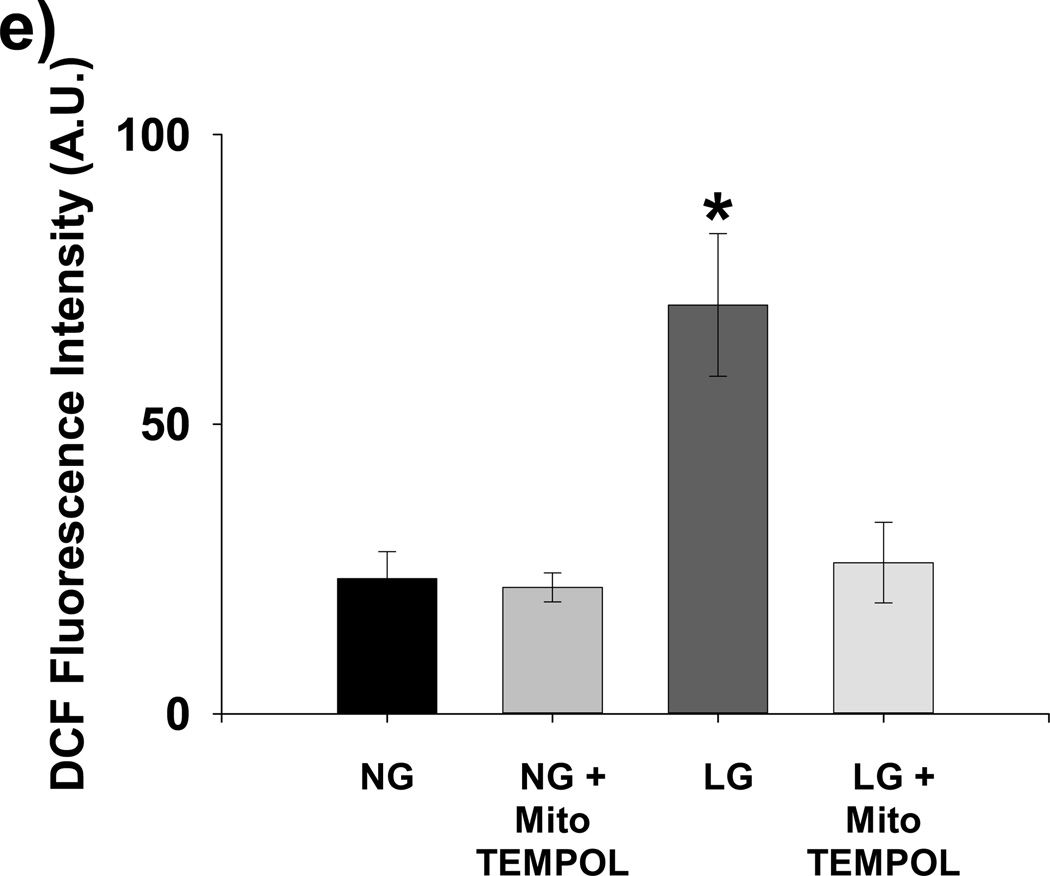
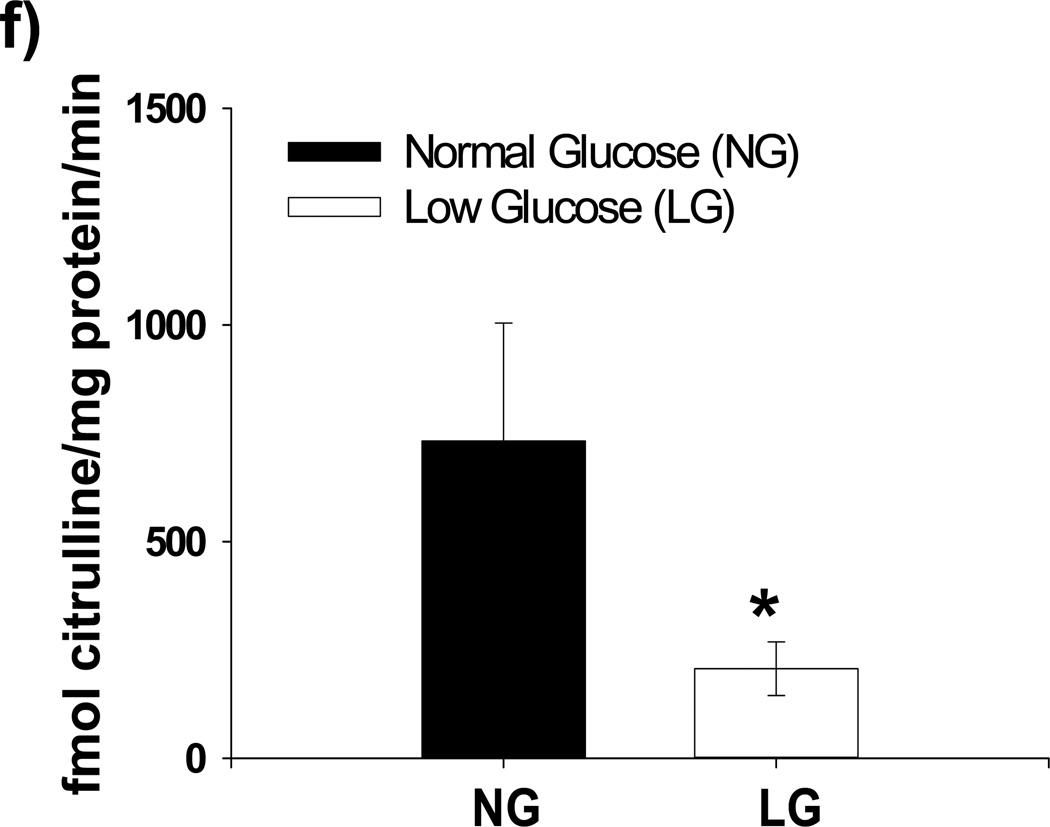
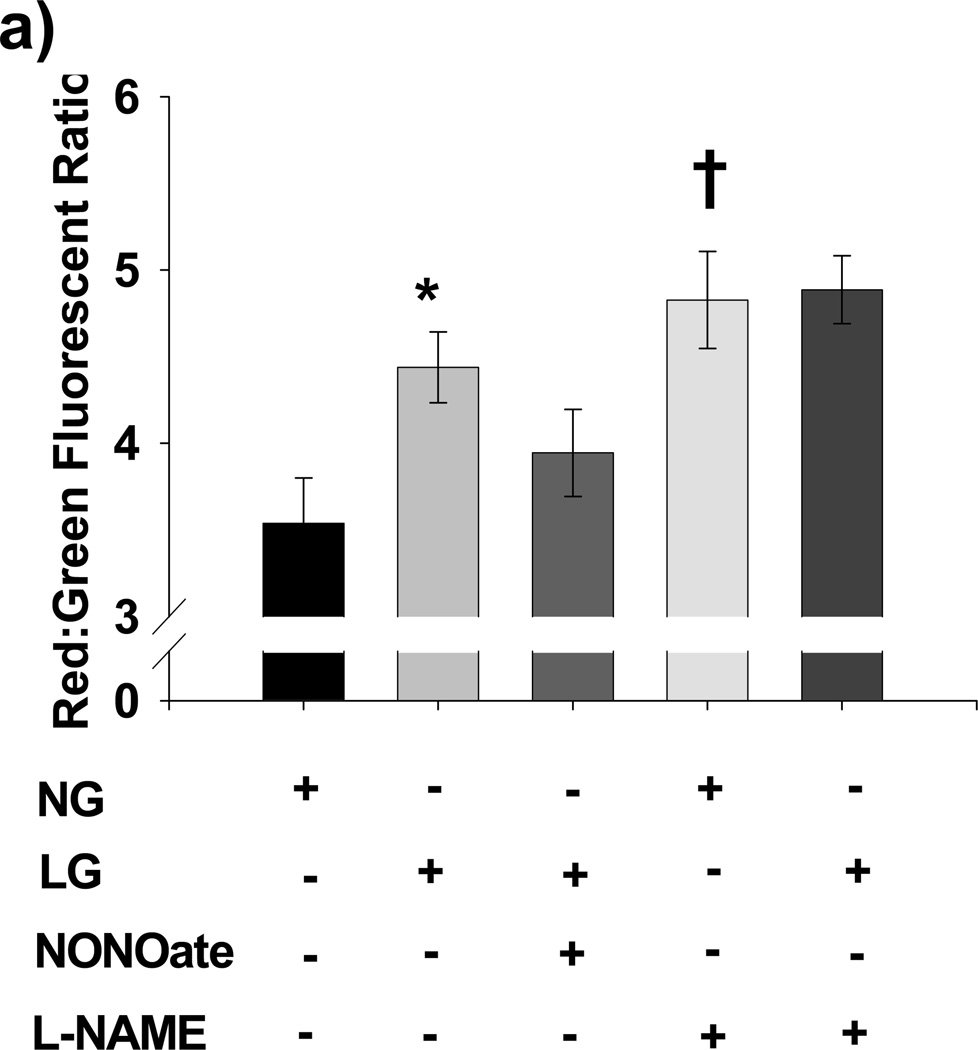
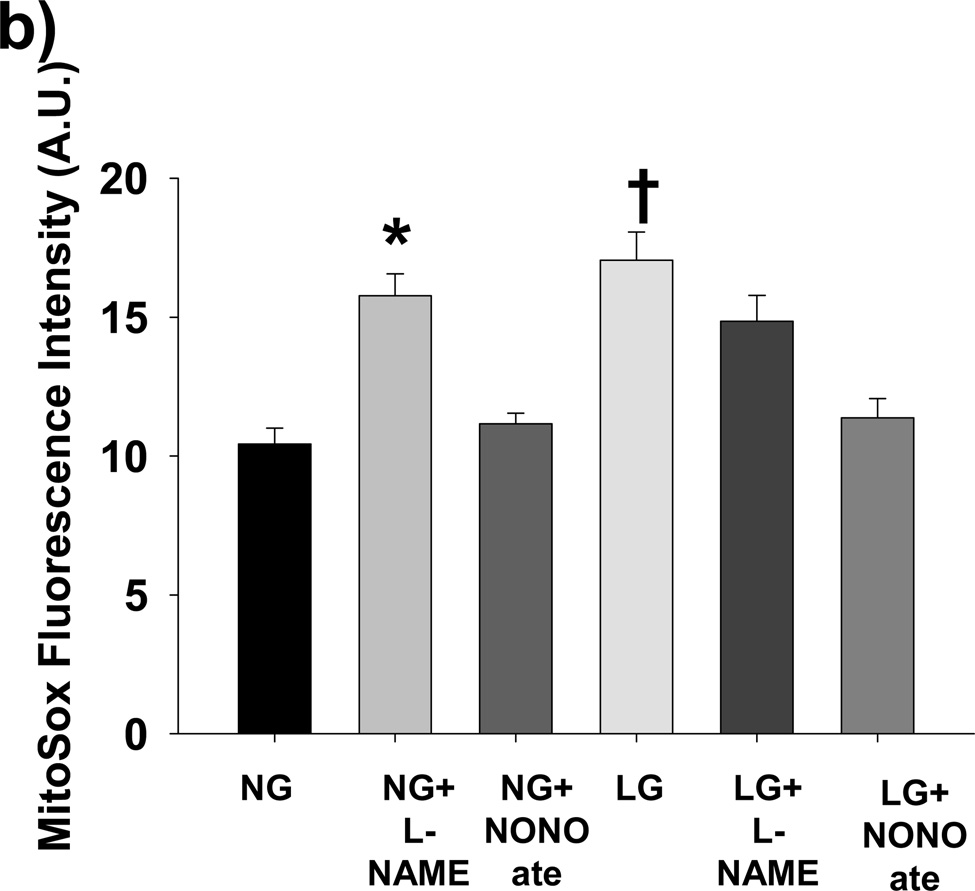
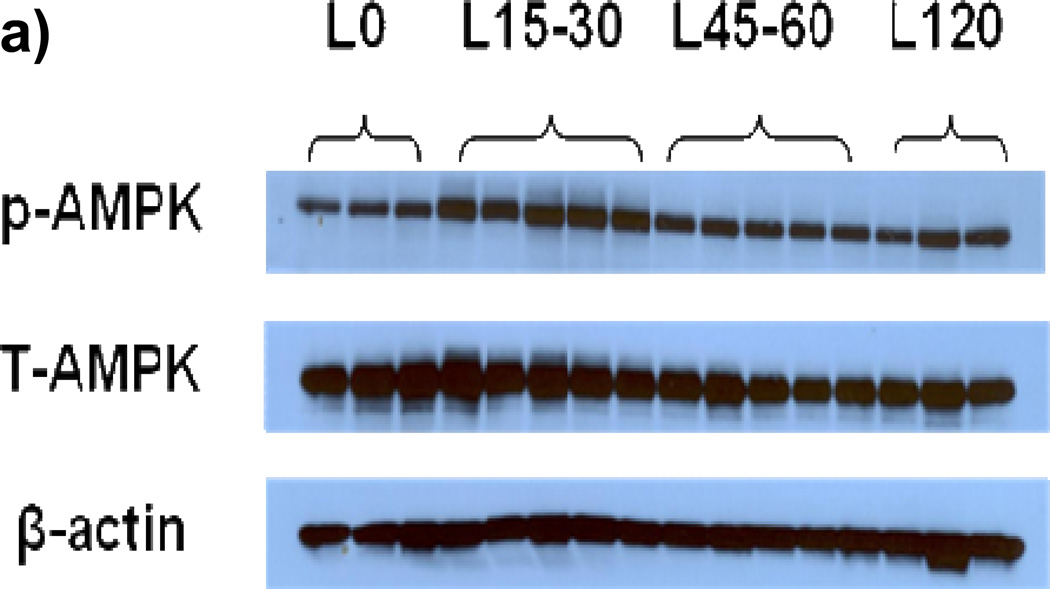
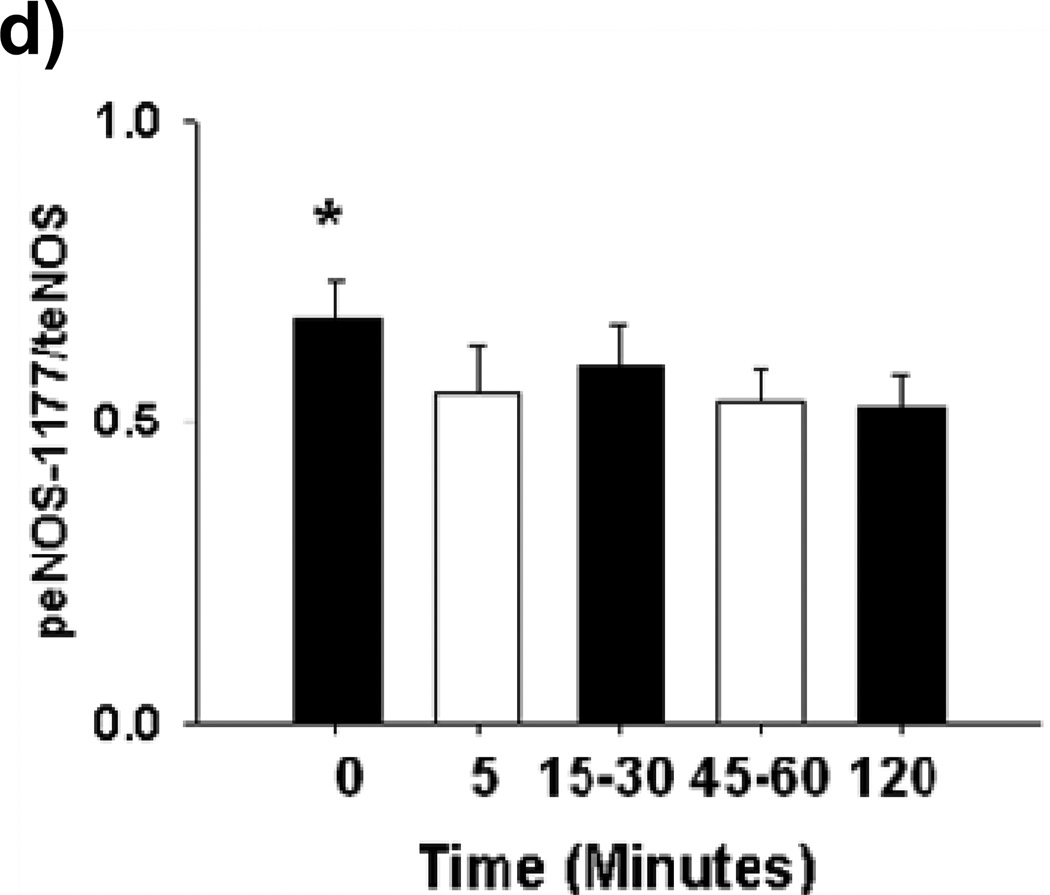
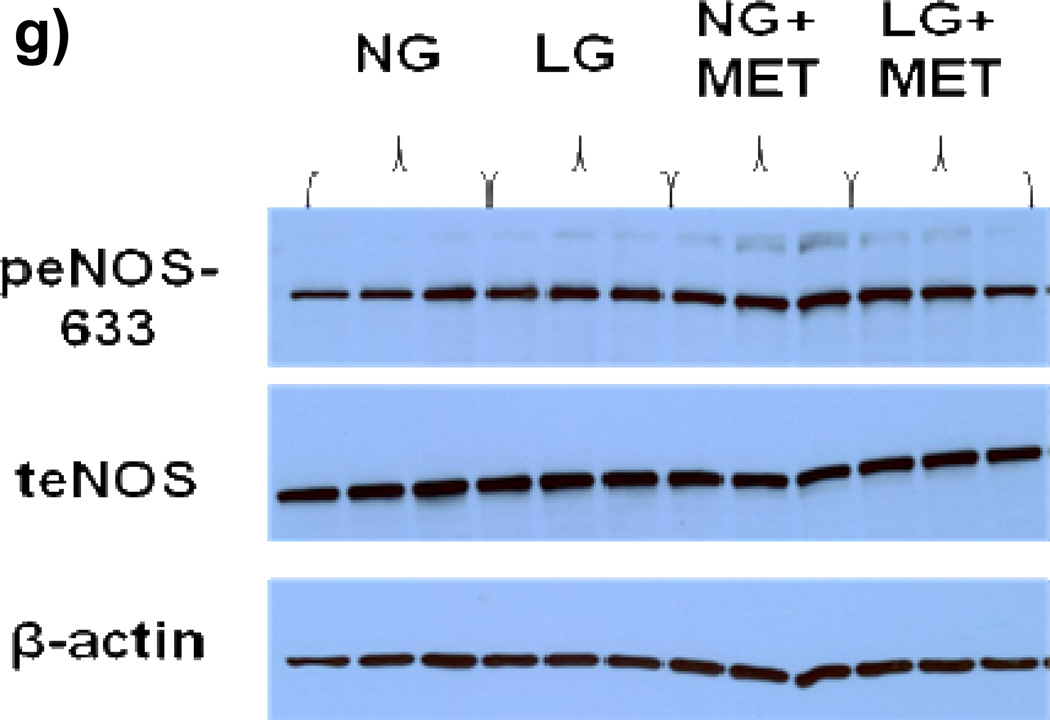
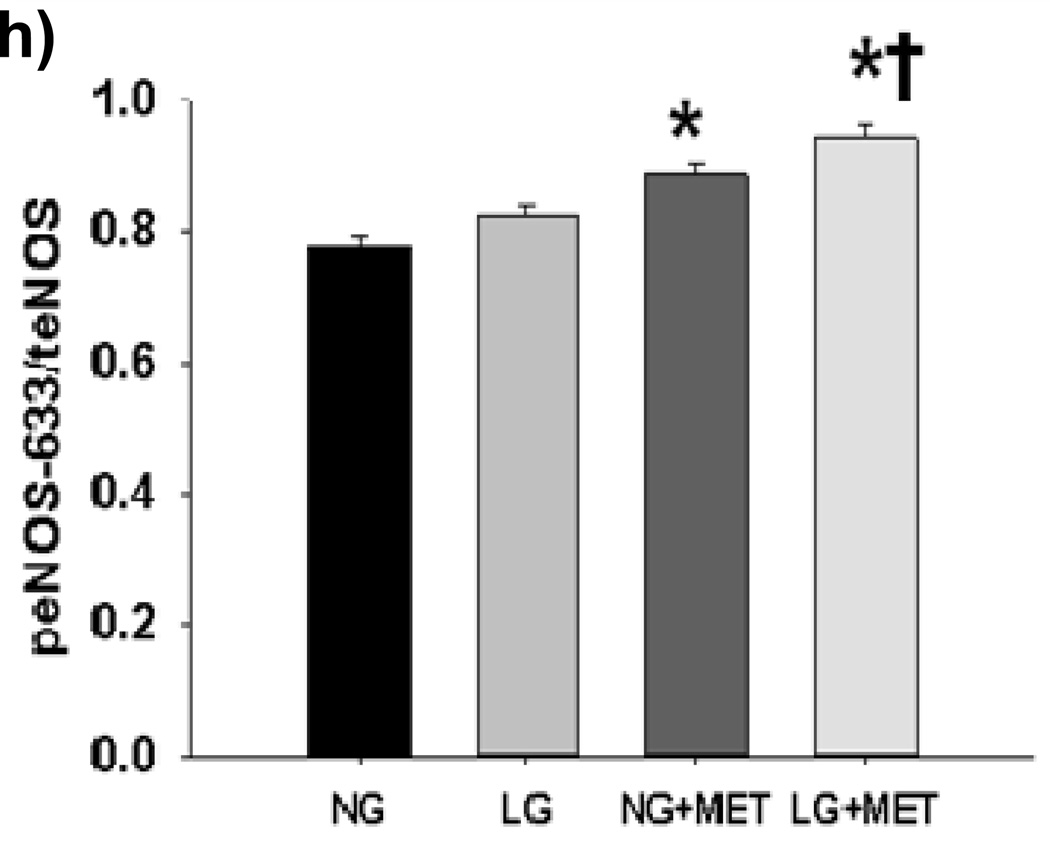
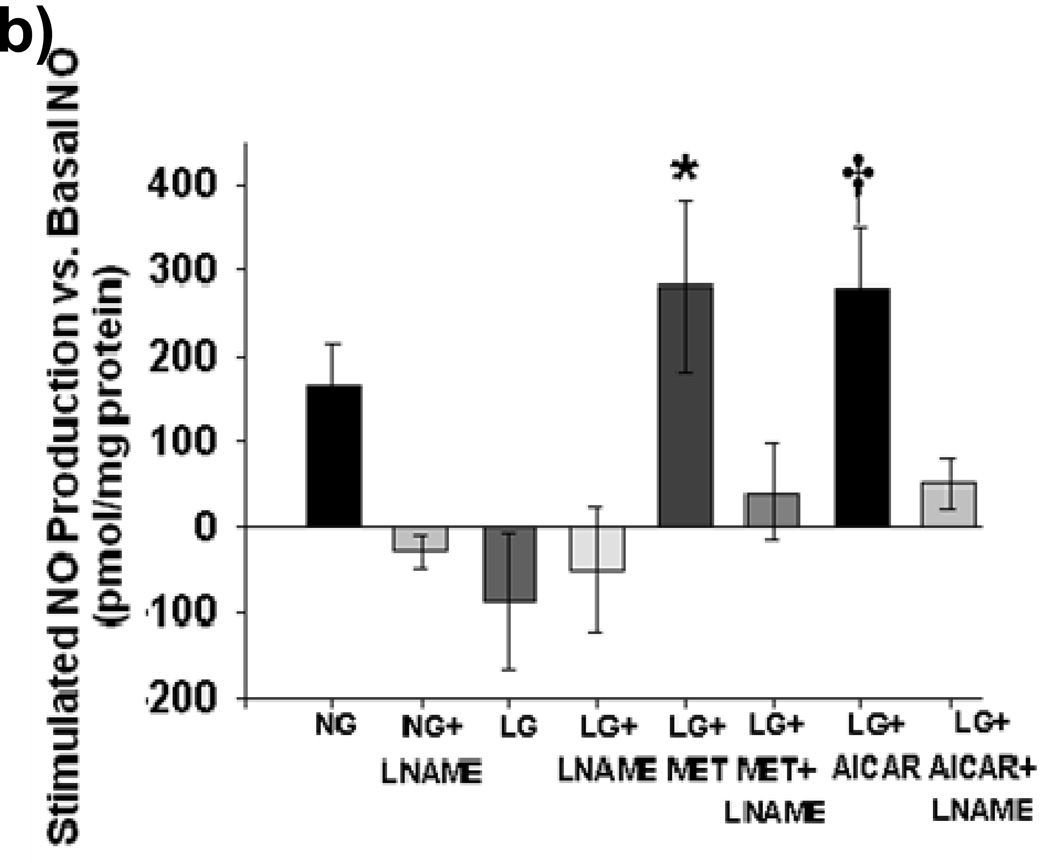
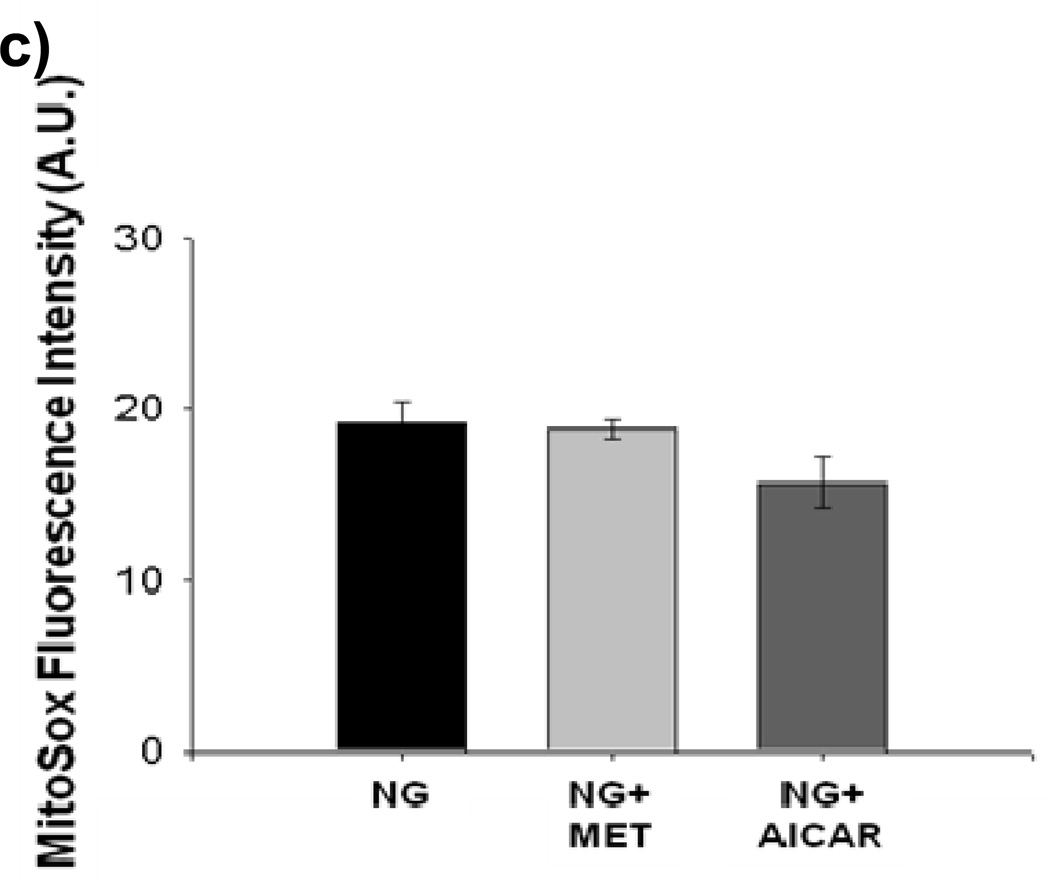
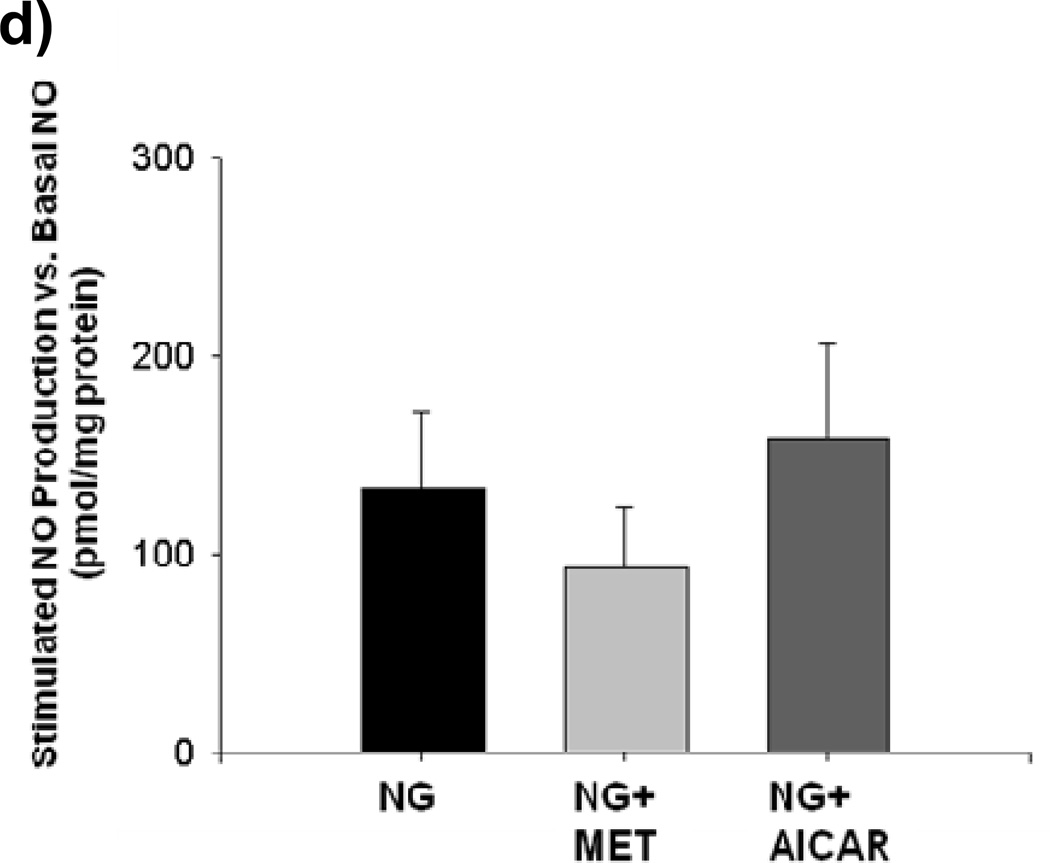
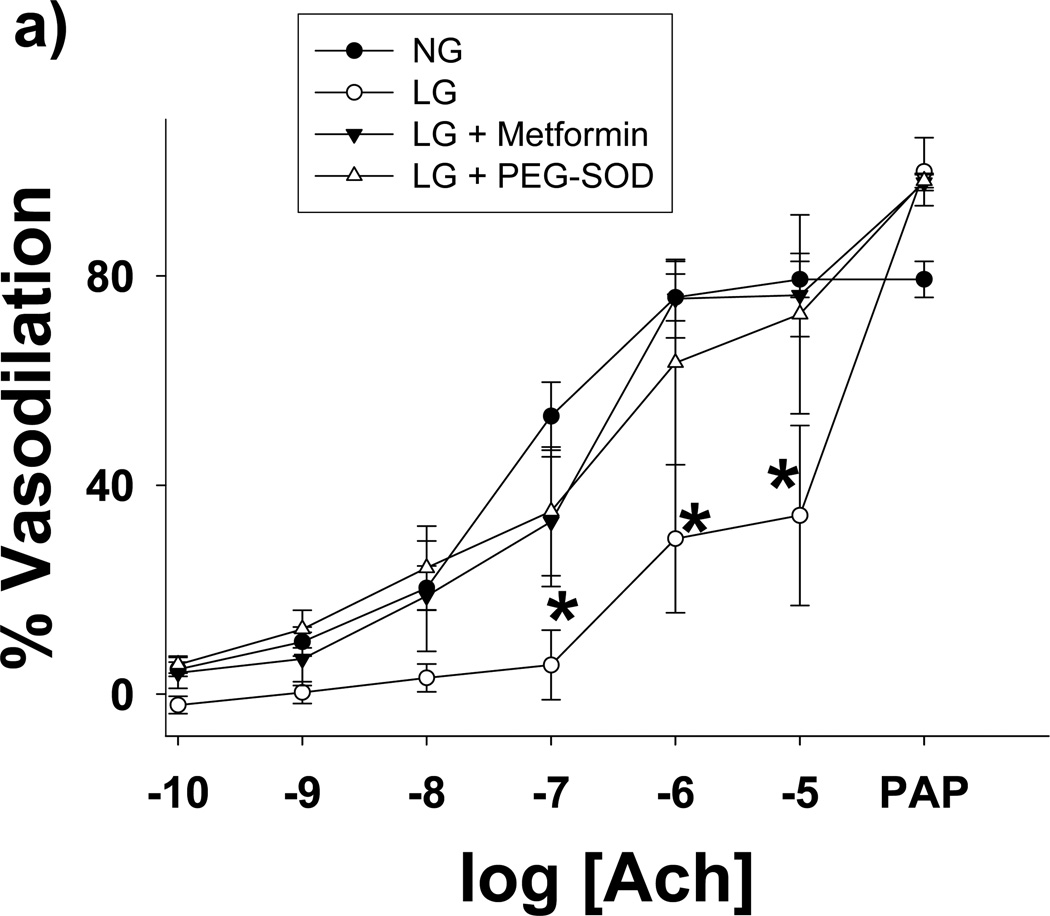
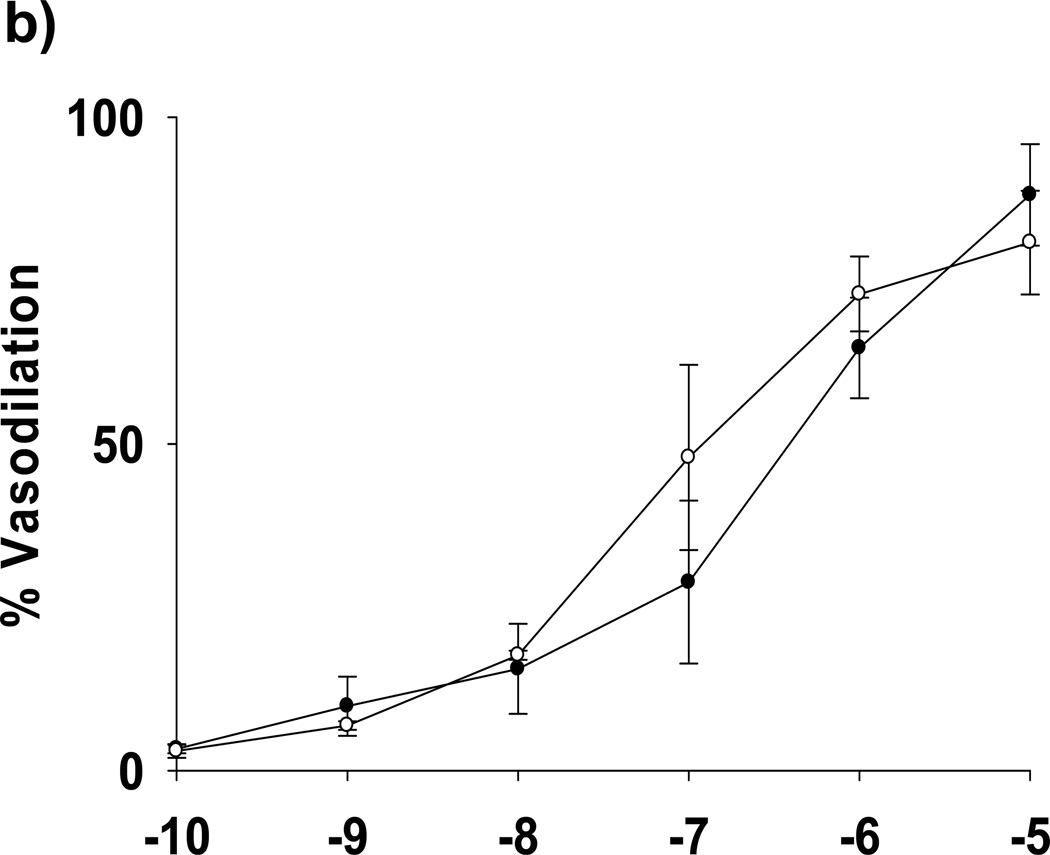
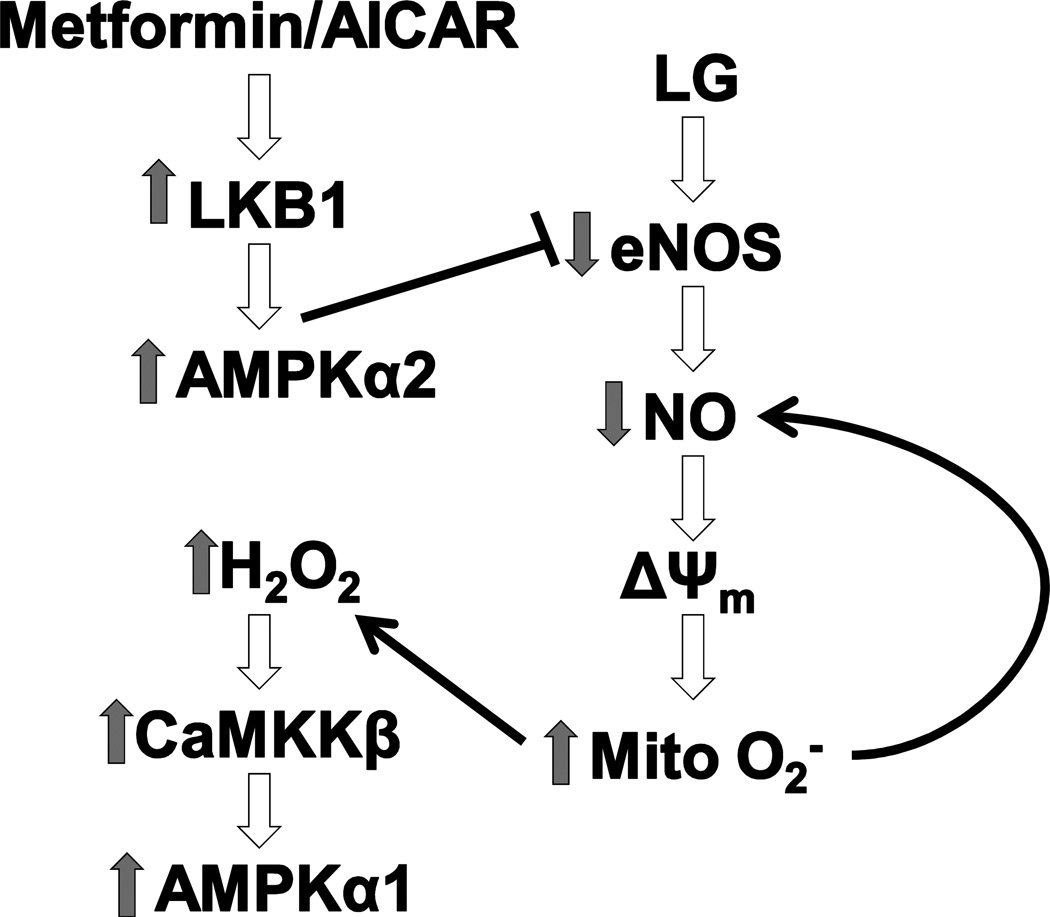
Methods and results: We exposed human umbilical vein endothelial cells to low glucose conditions in a clinically relevant range (40-70 mg/dL) and found rapid and marked reductions in nitric oxide (NO) bioavailability (P<0.001). This was associated with concomitantly increased mitochondrial superoxide production (P<0.001) and NO-dependent mitochondrial hyperpolarization (P<0.001). Reduced NO bioavailability was rapid and attributable to reduced endothelial nitric oxide synthase activity and destruction of NO. Low glucose rapidly activated AMP kinase, but physiological activation failed to restore NO bioavailability. Pharmacological AMP kinase activation led to phosphorylation of endothelial nitric oxide synthase's Ser633 activation site, reversing the adverse effects of low glucose. This protective effect was prevented by L-NG-Nitroarginine methyl ester. Intact human arterioles exposed to low glucose demonstrated marked endothelial dysfunction, which was prevented by either metformin or TEMPOL.
Conclusion: Our data suggest that moderate low glucose exposure rapidly impairs NO bioavailability and endothelial function in the human endothelium and that pharmacological AMP kinase activation inhibit this effect in an NO-dependent manner.
Figures








References
-
- Patel A, MacMahon S, Chalmers J, Neal B, Billot L, Woodward M, Marre M, Cooper M, Glasziou P, Grobbee D, Hamet P, Harrap S, Heller S, Liu L, Mancia G, Mogensen CE, Pan C, Poulter N, Rodgers A, Williams B, Bompoint S, de Galan BE, Joshi R, Travert F. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008;358:2560–2572. - PubMed
-
- Zoungas S, Patel A, Chalmers J, de Galan BE, Li Q, Billot L, Woodward M, Ninomiya T, Neal B, MacMahon S, Grobbee DE, Kengne AP, Marre M, Heller S. Severe hypoglycemia and risks of vascular events and death. N Engl J Med. 2010;363:1410–1418. - PubMed
-
- Finfer S, Chittock DR, Su SY, Blair D, Foster D, Dhingra V, Bellomo R, Cook D, Dodek P, Henderson WR, Hebert PC, Heritier S, Heyland DK, McArthur C, McDonald E, Mitchell I, Myburgh JA, Norton R, Potter J, Robinson BG, Ronco JJ. Intensive versus conventional glucose control in critically ill patients. N Engl J Med. 2009;360:1283–1297. - PubMed
-
- Kosiborod M, Inzucchi SE, Krumholz HM, Xiao L, Jones PG, Fiske S, Masoudi FA, Marso SP, Spertus JA. Glucometrics in patients hospitalized with acute myocardial infarction: defining the optimal outcomes-based measure of risk. Circulation. 2008;117:1018–1027. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
