

De novo and inherited mutations in COL4A2, encoding the type IV collagen α2 chain cause porencephaly
- PMID: 22209246
- PMCID: PMC3257897
- DOI: 10.1016/j.ajhg.2011.11.016
De novo and inherited mutations in COL4A2, encoding the type IV collagen α2 chain cause porencephaly
Abstract
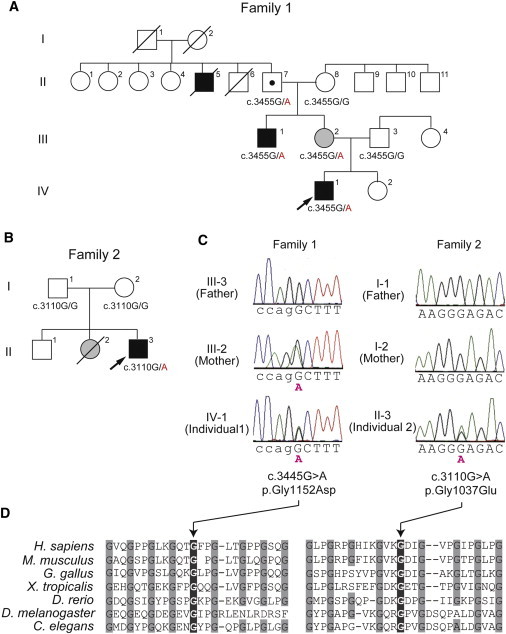
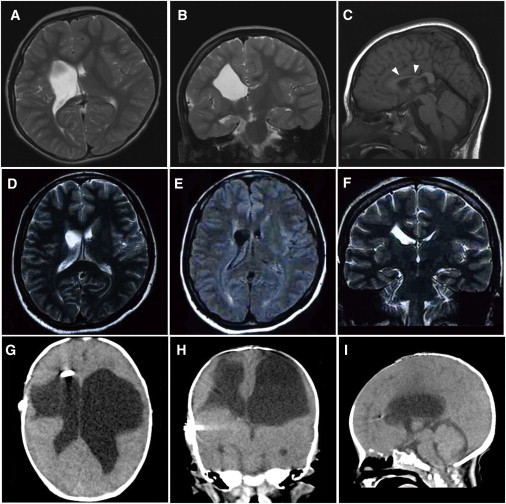
Porencephaly is a neurological disorder characterized by fluid-filled cysts or cavities in the brain that often cause hemiplegia. It has been suggested that porencephalic cavities result from focal cerebral degeneration involving hemorrhages. De novo or inherited heterozygous mutations in COL4A1, which encodes the type IV α1 collagen chain that is essential for structural integrity for vascular basement membranes, have been reported in individuals with porencephaly. Most mutations occurred at conserved Gly residues in the Gly-Xaa-Yaa repeats of the triple-helical domain, leading to alterations of the α1α1α2 heterotrimers. Here we report on two individuals with porencephaly caused by a heterozygous missense mutation in COL4A2, which encodes the type IV α2 collagen chain. Mutations c.3455G>A and c.3110G>A, one in each of the individuals, cause Gly residues in the Gly-Xaa-Yaa repeat to be substituted as p.Gly1152Asp and p.Gly1037Glu, respectively, probably resulting in alterations of the α1α1α2 heterotrimers. The c.3455G>A mutation was found in the proband's mother, who showed very mild monoparesis of the left upper extremity, and the maternal elder uncle, who had congenital hemiplegia. The maternal grandfather harboring the mutation is asymptomatic. The c.3110G>A mutation occurred de novo. Our study confirmed that abnormalities of the α1α1α2 heterotrimers of type IV collagen cause porencephaly and stresses the importance of screening for COL4A2 as well as for COL4A1.
Copyright © 2012 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
COL4A2 mutation associated with familial porencephaly and small-vessel disease.Eur J Hum Genet. 2012 Aug;20(8):844-51. doi: 10.1038/ejhg.2012.20. Epub 2012 Feb 15. Eur J Hum Genet. 2012. PMID: 22333902 Free PMC article.
-
Phenotypic spectrum of COL4A1 mutations: porencephaly to schizencephaly.Ann Neurol. 2013 Jan;73(1):48-57. doi: 10.1002/ana.23736. Epub 2012 Dec 7. Ann Neurol. 2013. PMID: 23225343
-
Chemical chaperone treatment reduces intracellular accumulation of mutant collagen IV and ameliorates the cellular phenotype of a COL4A2 mutation that causes haemorrhagic stroke.Hum Mol Genet. 2014 Jan 15;23(2):283-92. doi: 10.1093/hmg/ddt418. Epub 2013 Sep 2. Hum Mol Genet. 2014. PMID: 24001601 Free PMC article.
-
The expanding phenotype of COL4A1 and COL4A2 mutations: clinical data on 13 newly identified families and a review of the literature.Genet Med. 2015 Nov;17(11):843-53. doi: 10.1038/gim.2014.210. Epub 2015 Feb 26. Genet Med. 2015. PMID: 25719457 Review.
-
Childhood presentation of COL4A1 mutations.Dev Med Child Neurol. 2012 Jun;54(6):569-74. doi: 10.1111/j.1469-8749.2011.04198.x. Epub 2012 Jan 16. Dev Med Child Neurol. 2012. PMID: 22574627 Review.
Cited by
-
Intracerebral Hemorrhage Genetics.Genes (Basel). 2022 Jul 15;13(7):1250. doi: 10.3390/genes13071250. Genes (Basel). 2022. PMID: 35886033 Free PMC article. Review.
-
The triple helix of collagens - an ancient protein structure that enabled animal multicellularity and tissue evolution.J Cell Sci. 2018 Apr 9;131(7):jcs203950. doi: 10.1242/jcs.203950. J Cell Sci. 2018. PMID: 29632050 Free PMC article. Review.
-
Elevated TGFβ signaling contributes to cerebral small vessel disease in mouse models of Gould syndrome.Matrix Biol. 2023 Jan;115:48-70. doi: 10.1016/j.matbio.2022.11.007. Epub 2022 Nov 23. Matrix Biol. 2023. PMID: 36435425 Free PMC article.
-
Psychosis Associated with Acquired Porencephaly-Cause or Incidental Finding? Case Report and Review of Literature.Medicina (Kaunas). 2022 Apr 24;58(5):586. doi: 10.3390/medicina58050586. Medicina (Kaunas). 2022. PMID: 35630003 Free PMC article. Review.
-
Heterozygous Pathogenic and Likely Pathogenic Symptomatic HTRA1 Variant Carriers in Cerebral Small Vessel Disease.Int J Gen Med. 2023 Mar 29;16:1149-1162. doi: 10.2147/IJGM.S404813. eCollection 2023. Int J Gen Med. 2023. PMID: 37016629 Free PMC article. Review.
References
-
- Berg R.A., Aleck K.A., Kaplan A.M. Familial porencephaly. Arch. Neurol. 1983;40:567–569. - PubMed
-
- Govaert P. Prenatal stroke. Semin. Fetal Neonatal Med. 2009;14:250–266. - PubMed
-
- Hunter A. In: Human Malformations and related Anomalies. Re S., Jg H., editors. Oxford University Press; New York: 2006. Porencephaly; pp. 645–654.
-
- Mancini G.M., de Coo I.F., Lequin M.H., Arts W.F. Hereditary porencephaly: clinical and MRI findings in two Dutch families. Eur. J. Paediatr. Neurol. 2004;8:45–54. - PubMed
-
- Vilain C., Van Regemorter N., Verloes A., David P., Van Bogaert P. Neuroimaging fails to identify asymptomatic carriers of familial porencephaly. Am. J. Med. Genet. 2002;112:198–202. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
