

The NF-κB subunit c-Rel stimulates cardiac hypertrophy and fibrosis
- PMID: 22210479
- PMCID: PMC3314915
- DOI: 10.1016/j.ajpath.2011.11.007
The NF-κB subunit c-Rel stimulates cardiac hypertrophy and fibrosis
Abstract
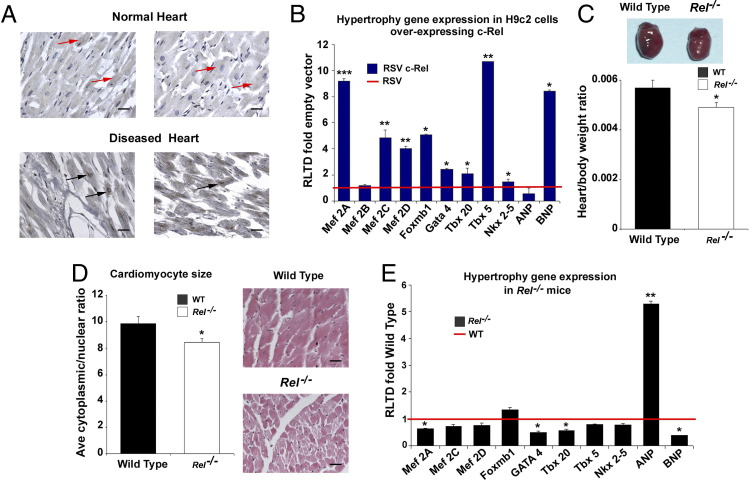
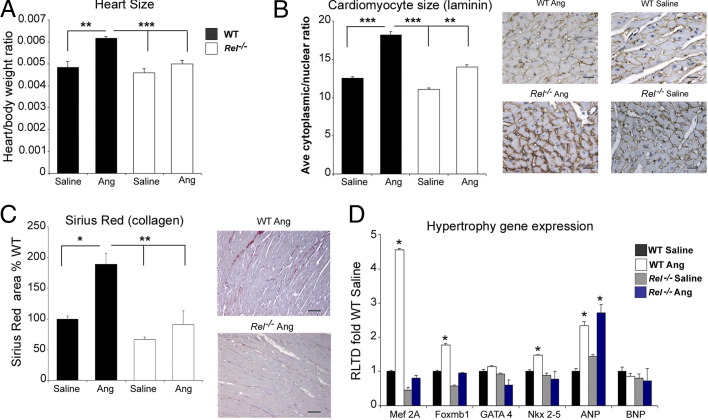
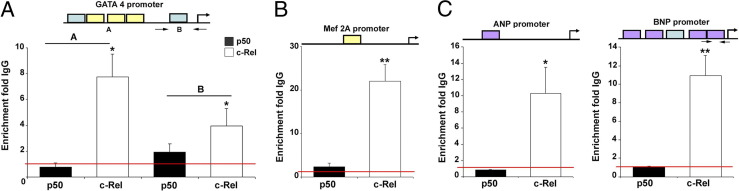
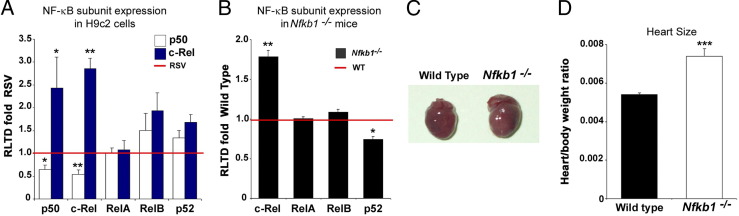
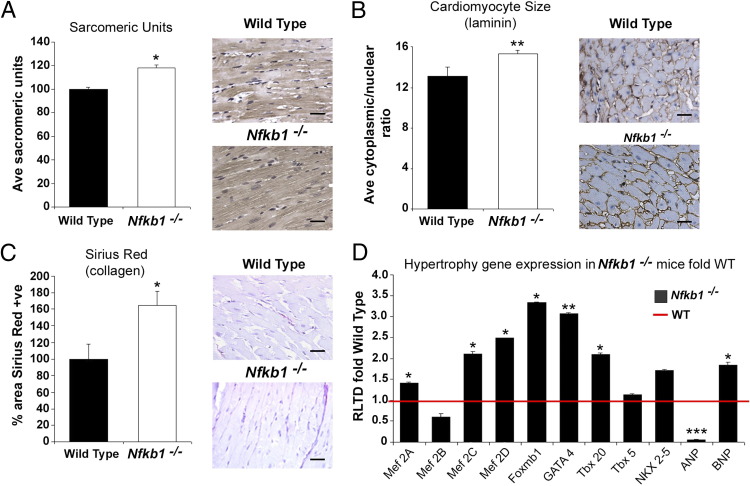
Cardiac remodeling and hypertrophy are the pathological consequences of cardiovascular disease and are correlated with its associated mortality. Activity of the transcription factor NF-κB is increased in the diseased heart; however, our present understanding of how the individual subunits contribute to cardiovascular disease is limited. We assign a new role for the c-Rel subunit as a stimulator of cardiac hypertrophy and fibrosis. We discovered that c-Rel-deficient mice have smaller hearts at birth, as well as during adulthood, and are protected from developing cardiac hypertrophy and fibrosis after chronic angiotensin infusion. Results of both gene expression and cross-linked chromatin immunoprecipitation assay analyses identified transcriptional activators of hypertrophy, myocyte enhancer family, Gata4, and Tbx proteins as Rel gene targets. We suggest that the p50 subunit could limit the prohypertrophic actions of c-Rel in the normal heart, because p50 overexpression in H9c2 cells repressed c-Rel levels and the absence of cardiac p50 was associated with increases in both c-Rel levels and cardiac hypertrophy. We report for the first time that c-Rel is highly expressed and confined to the nuclei of diseased adult human hearts but is restricted to the cytoplasm of normal cardiac tissues. We conclude that c-Rel-dependent signaling is critical for both cardiac remodeling and hypertrophy. Targeting its activities could offer a novel therapeutic strategy to limit the effects of cardiac disease.
Copyright © 2012 American Society for Investigative Pathology. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Targeted deletion of nuclear factor kappaB p50 enhances cardiac remodeling and dysfunction following myocardial infarction.Circ Res. 2009 Mar 13;104(5):699-706. doi: 10.1161/CIRCRESAHA.108.189746. Epub 2009 Jan 24. Circ Res. 2009. PMID: 19168865
-
NF-kappaB/p50 and NF-kappaB/c-Rel differentially regulate the activity of the 3'alphaE-hsl,2 enhancer in normal murine B cells in an activation-dependent manner.Int Immunol. 2000 Aug;12(8):1167-72. doi: 10.1093/intimm/12.8.1167. Int Immunol. 2000. PMID: 10917891
-
The c-Rel subunit of NF-κB regulates epidermal homeostasis and promotes skin fibrosis in mice.Am J Pathol. 2013 Jun;182(6):2109-20. doi: 10.1016/j.ajpath.2013.02.016. Epub 2013 Apr 4. Am J Pathol. 2013. PMID: 23562440 Free PMC article.
-
New insights into the roles of ReL/NF-kappa B transcription factors in immune function, hemopoiesis and human disease.Int J Biochem Cell Biol. 1999 Oct;31(10):1209-19. doi: 10.1016/s1357-2725(99)00068-0. Int J Biochem Cell Biol. 1999. PMID: 10582348 Review.
-
The c-Rel transcription factor and B-cell proliferation: a deal with the devil.Oncogene. 2004 Mar 25;23(13):2275-86. doi: 10.1038/sj.onc.1207410. Oncogene. 2004. PMID: 14755244 Review.
Cited by
-
Association of rheumatoid arthritis susceptibility gene with lipid profiles in patients with rheumatoid arthritis.Rheumatology (Oxford). 2014 Jun;53(6):1014-21. doi: 10.1093/rheumatology/ket472. Rheumatology (Oxford). 2014. PMID: 24489016 Free PMC article.
-
Exosomes: From Potential Culprits to New Therapeutic Promise in the Setting of Cardiac Fibrosis.Cells. 2020 Mar 2;9(3):592. doi: 10.3390/cells9030592. Cells. 2020. PMID: 32131460 Free PMC article. Review.
-
Reactive Oxygen Species Related Noncoding RNAs as Regulators of Cardiovascular Diseases.Int J Biol Sci. 2019 Jan 24;15(3):680-687. doi: 10.7150/ijbs.30464. eCollection 2019. Int J Biol Sci. 2019. PMID: 30745854 Free PMC article. Review.
-
c-Rel orchestrates energy-dependent epithelial and macrophage reprogramming in fibrosis.Nat Metab. 2020 Nov;2(11):1350-1367. doi: 10.1038/s42255-020-00306-2. Epub 2020 Nov 9. Nat Metab. 2020. PMID: 33168981 Free PMC article.
-
Preoperative myocardial expression of E3 ubiquitin ligases in aortic stenosis patients undergoing valve replacement and their association to postoperative hypertrophy.PLoS One. 2020 Sep 18;15(9):e0237000. doi: 10.1371/journal.pone.0237000. eCollection 2020. PLoS One. 2020. PMID: 32946439 Free PMC article.
References
-
- Franco M., Cooper R.S., Bilal U., Fuster V. Challenges and opportunities for cardiovascular disease prevention. Am J Med. 2011;124:95–102. - PubMed
-
- Barry S.P., Davidson S.M., Townsend P.A. Molecular regulation of cardiac hypertrophy. Int J Biochem Cell Biol. 2008;40:2023–2039. - PubMed
-
- Chien K.R. Stress pathways and heart failure. Cell. 1999;98:555–558. - PubMed
-
- Frey N., Olson E.N. Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol. 2003;65:45–79. - PubMed
-
- Chevalier B., Callens-el Amrani F., Heymes C., Swynghedauw B. Molecular basis of regression of cardiac hypertrophy. Am J Cardiol. 1994;73:10C–17C. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
