

Crystal structure of human polynucleotide phosphorylase: insights into its domain function in RNA binding and degradation
- PMID: 22210891
- PMCID: PMC3351181
- DOI: 10.1093/nar/gkr1281
Crystal structure of human polynucleotide phosphorylase: insights into its domain function in RNA binding and degradation
Abstract
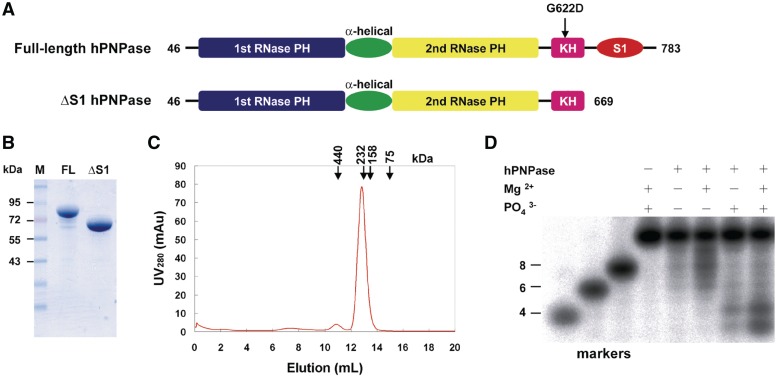
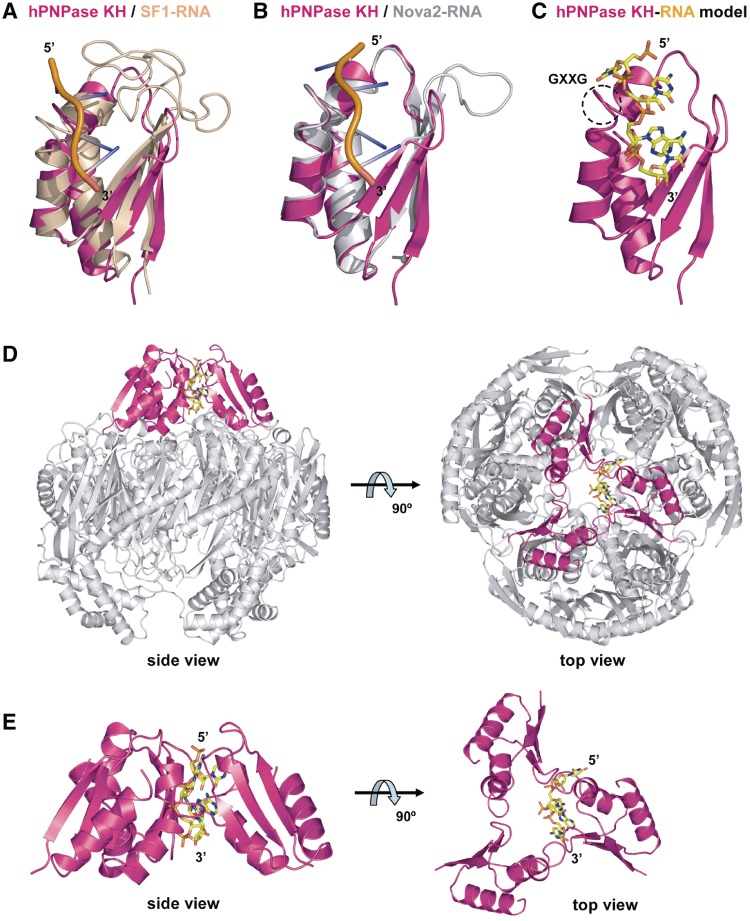
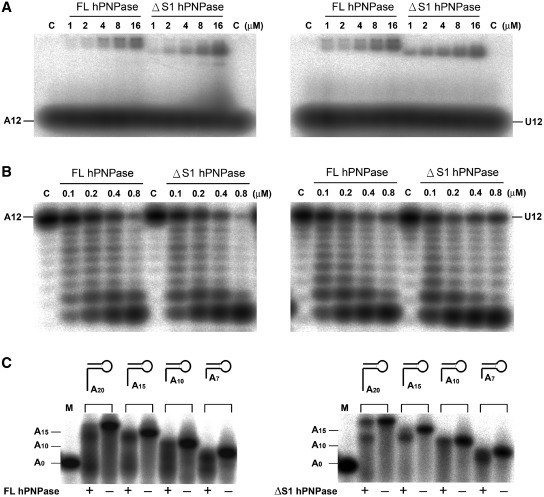
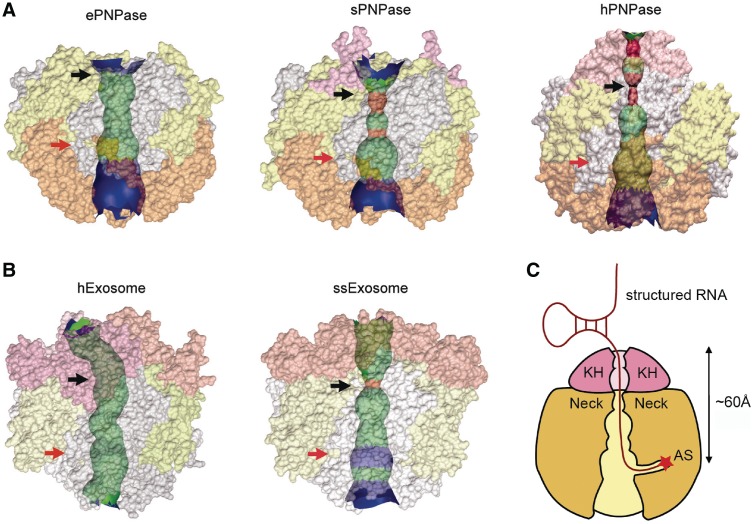
Human polynucleotide phosphorylase (hPNPase) is a 3'-to-5' exoribonuclease that degrades specific mRNA and miRNA, and imports RNA into mitochondria, and thus regulates diverse physiological processes, including cellular senescence and homeostasis. However, the RNA-processing mechanism by hPNPase, particularly how RNA is bound via its various domains, remains obscure. Here, we report the crystal structure of an S1 domain-truncated hPNPase at a resolution of 2.1 Å. The trimeric hPNPase has a hexameric ring-like structure formed by six RNase PH domains, capped with a trimeric KH pore. Our biochemical and mutagenesis studies suggest that the S1 domain is not critical for RNA binding, and conversely, that the conserved GXXG motif in the KH domain directly participates in RNA binding in hPNPase. Our studies thus provide structural and functional insights into hPNPase, which uses a KH pore to trap a long RNA 3' tail that is further delivered into an RNase PH channel for the degradation process. Structural RNA with short 3' tails are, on the other hand, transported but not digested by hPNPase.
Figures



References
-
- Andrade JM, Pobre V, Silva IJ, Domingues S, Arraiano CM. The role of 3′–5′ exoribonucleases in RNA degradation. Prog. Mol. Biol. Trans. Sci. 2009;85:187–229. - PubMed
-
- Marcaida MJ, DePristo MA, Chandran V, Carpousis AJ, Luisi BF. The RNA degradosome: life in the fast lane of adaptive molecular evolution. Trends Biochem. Sci. 2006;31:359–365. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
