

TLR2 activation enhances HIV nuclear import and infection through T cell activation-independent and -dependent pathways
- PMID: 22210918
- PMCID: PMC3262879
- DOI: 10.4049/jimmunol.1102098
TLR2 activation enhances HIV nuclear import and infection through T cell activation-independent and -dependent pathways
Abstract
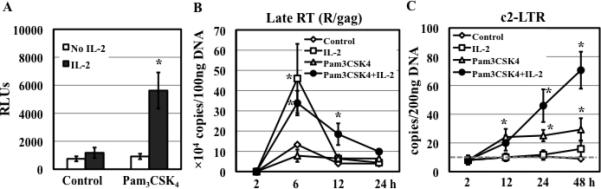
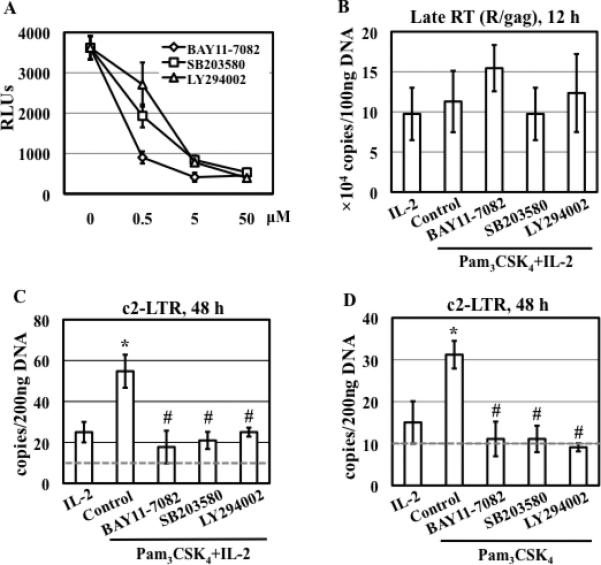
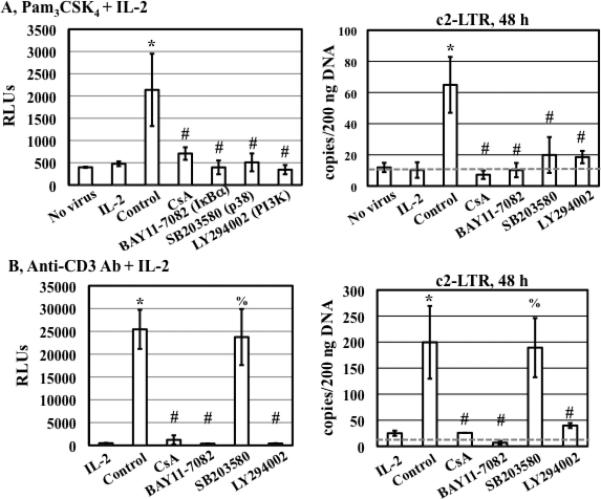
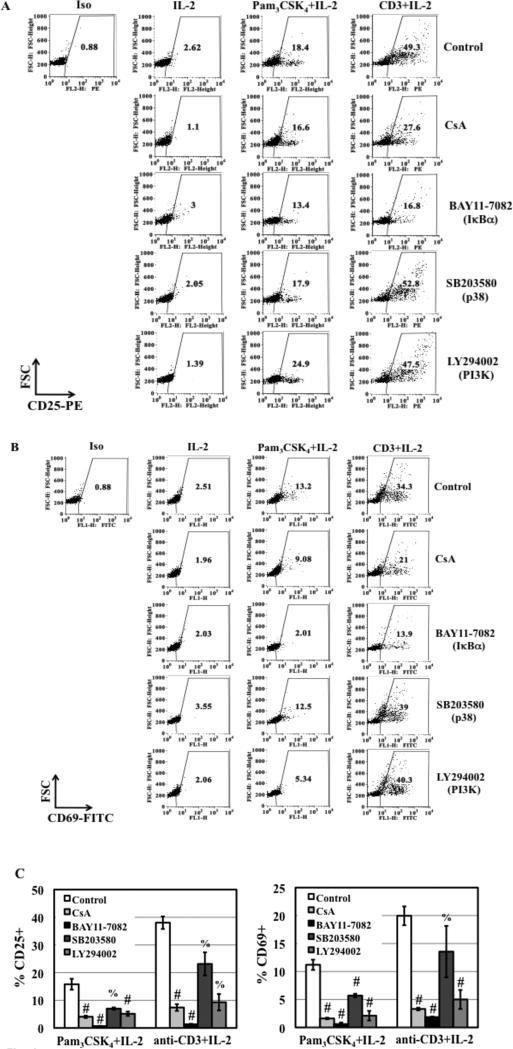
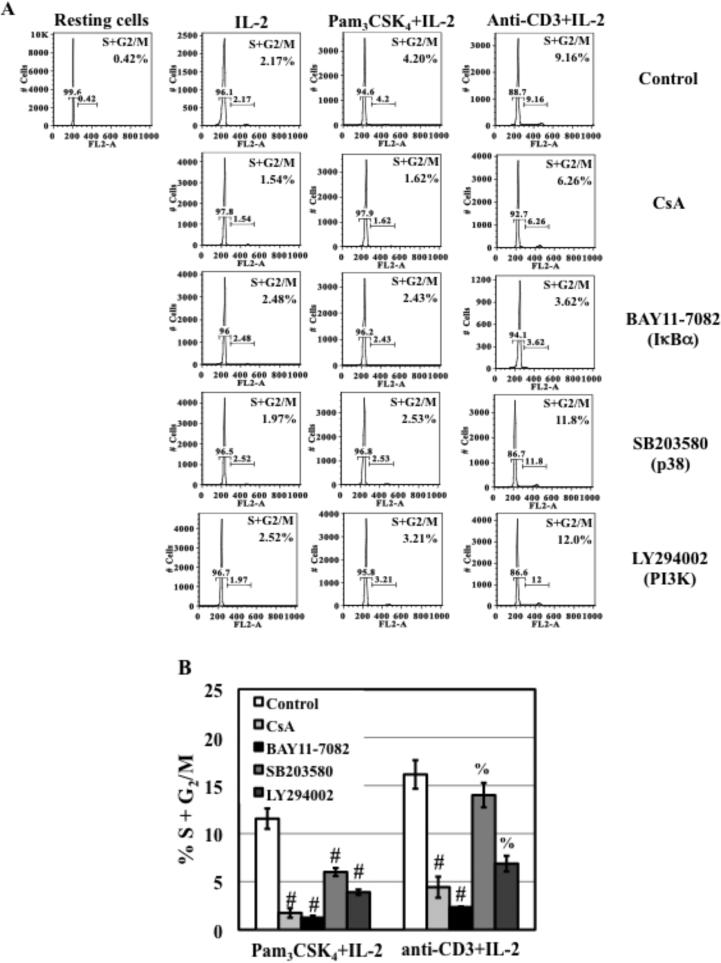
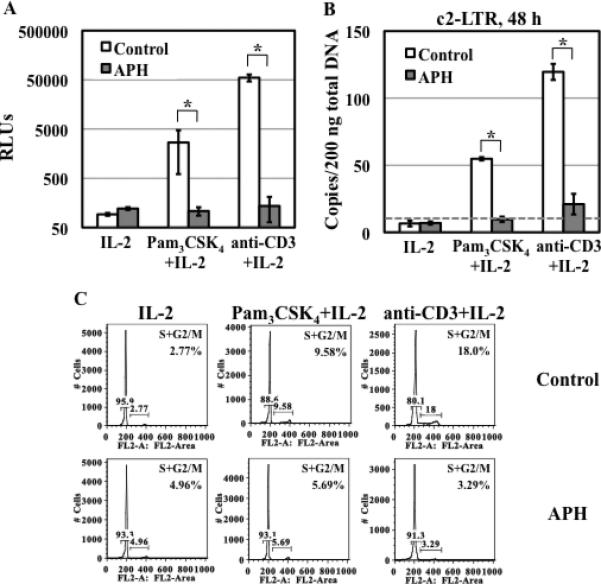
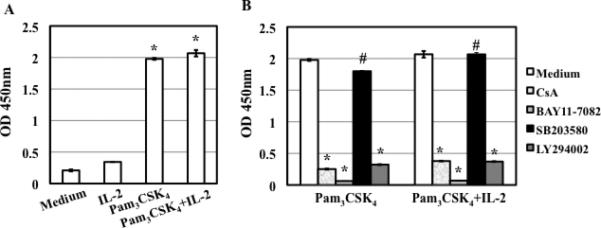
TLR2 activation plays a crucial role in Neisseria gonorrheae-mediated enhancement of HIV infection of resting CD4(+) T cells. We examined signaling pathways involved in the HIV enhancing effect of TLR2. TLR2 but not IL-2 signals promoted HIV nuclear import; however, both signals were required for the maximal enhancing effect. Although TLR2 signaling could not activate T cells, it increased IL-2-induced T cell activation. Cyclosporin A and IkBα inhibitor blocked TLR2-mediated enhancement of HIV infection/nuclear import. PI3K inhibitor blocked HIV infection/nuclear import and T cell activation and exerted a moderate inhibitory effect on cell cycle progression in CD4(+) T cells activated by TLR2/IL-2. Blockade of p38 signaling suppressed TLR2-mediated enhancement of HIV nuclear import/infection. However, the p38 inhibitor did not have a significant effect on T cell activation or TCR/CD3-mediated enhancement of HIV infection/nuclear import. The cell cycle arresting reagent aphidicolin blocked TLR2- and TCR/CD3-induced HIV infection/nuclear import. Finally, cyclosporin A and IκBα and PI3K inhibitors but not the p38 inhibitor blocked TLR2-mediated IκBα phosphorylation. Our results suggest that TLR2 activation enhances HIV infection/nuclear import in resting CD4(+) T cells through both T cell activation-dependent and -independent mechanisms.
Figures
References
-
- Cohen MS, Miller WC. Sexually transmitted diseases and human immunodeficiency virus infection: cause, effect, or both? Int J Infect Dis. 1998;3:1–4. - PubMed
-
- Plummer FA, Simonsen JN, Cameron DW, Ndinya-Achola JO, Kreiss JK, Gakinya MN, Waiyaki P, Cheang M, Piot P, Ronald AR, et al. Cofactors in male-female sexual transmission of human immunodeficiency virus type 1. J Infect Dis. 1991;163:233–239. - PubMed
-
- Cameron DW, Simonsen JN, D'Costa LJ, Ronald AR, Maitha GM, Gakinya MN, Cheang M, Ndinya-Achola JO, Piot P, Brunham RC, et al. Female to male transmission of human immunodeficiency virus type 1: risk factors for seroconversion in men. Lancet. 1989;2:403–407. - PubMed
-
- Ghys PD, Fransen K, Diallo MO, Ettiegne-Traore V, Coulibaly IM, Yeboue KM, Kalish ML, Maurice C, Whitaker JP, Greenberg AE, Laga M. The associations between cervicovaginal HIV shedding, sexually transmitted diseases and immunosuppression in female sex workers in Abidjan, Cote d'Ivoire. AIDS. 1997;11:F85–93. - PubMed
-
- Prebeck S, Brade H, Kirschning CJ, da Costa CP, Durr S, Wagner H, Miethke T. The Gram-negative bacterium Chlamydia trachomatis L2 stimulates tumor necrosis factor secretion by innate immune cells independently of its endotoxin. Microbes Infect. 2003;5:463–470. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
