

A distinct response to endogenous DNA damage in the development of Nbs1-deficient cortical neurons
- PMID: 22212482
- PMCID: PMC3343649
- DOI: 10.1038/cr.2012.3
A distinct response to endogenous DNA damage in the development of Nbs1-deficient cortical neurons
Abstract
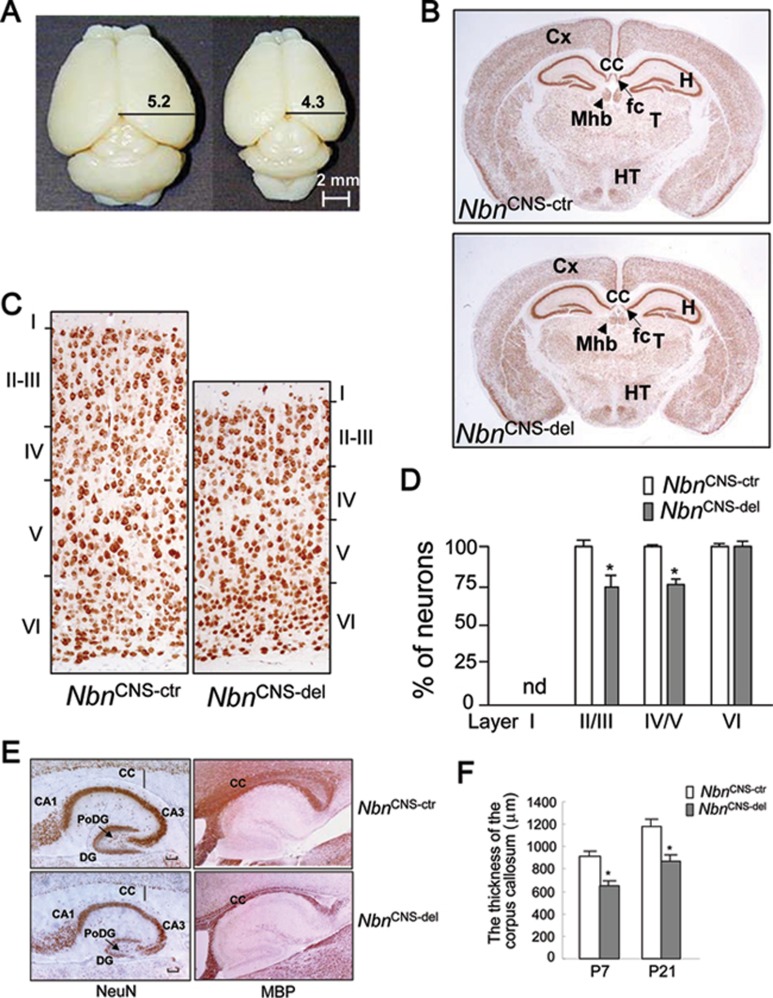
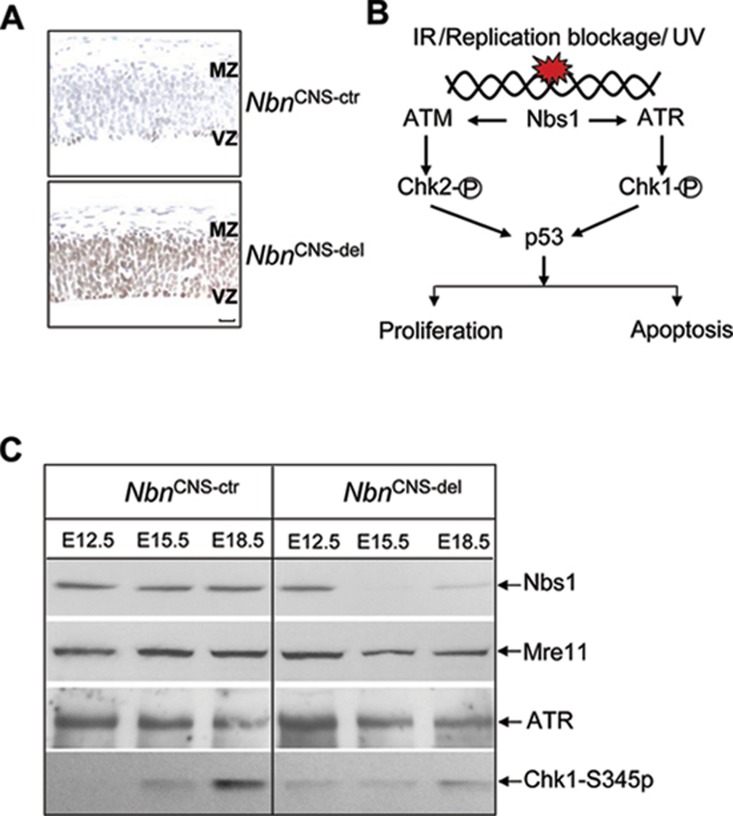
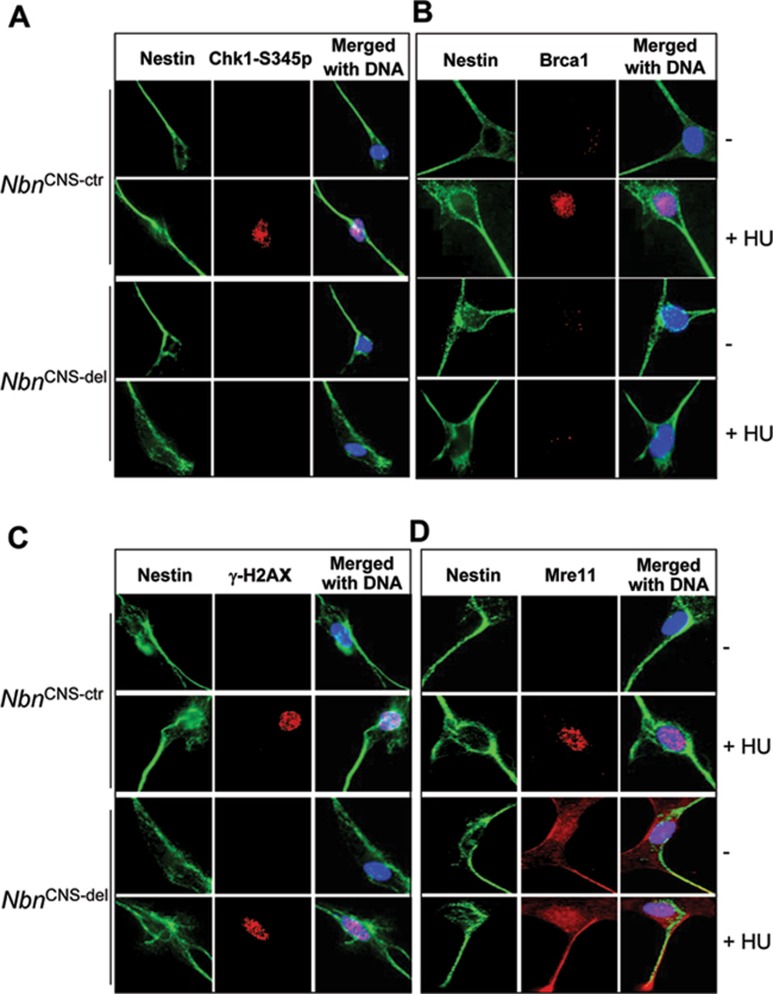
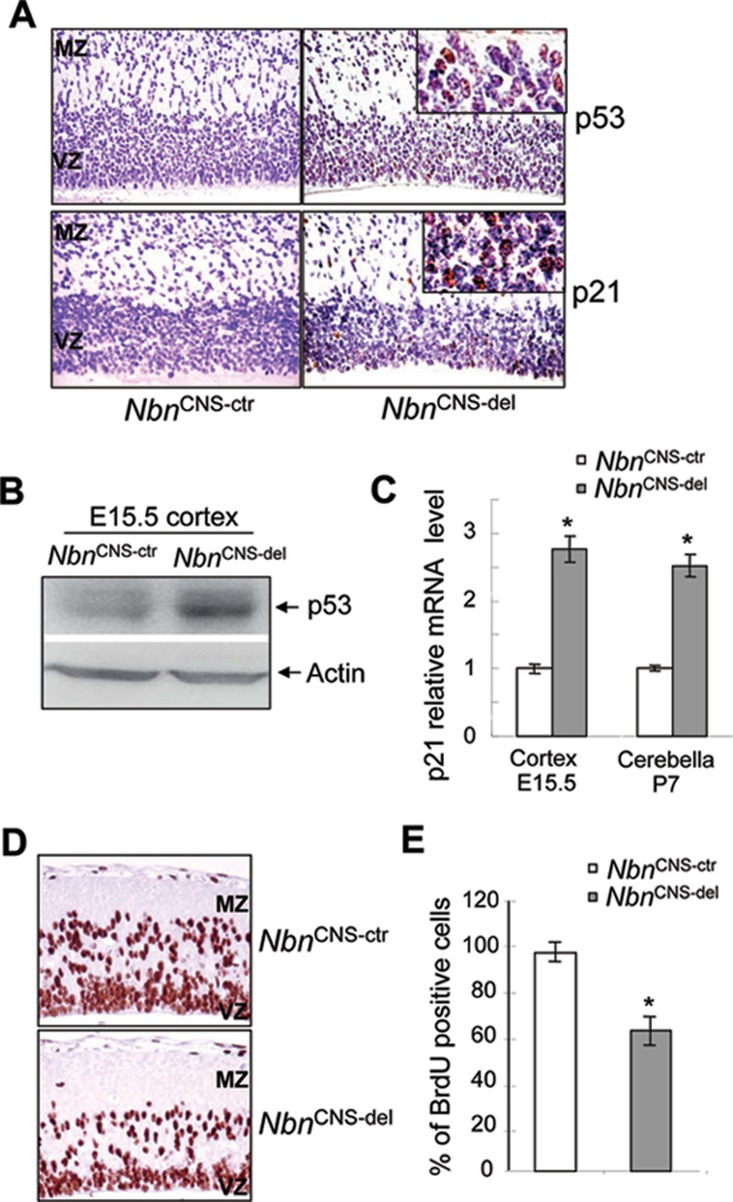
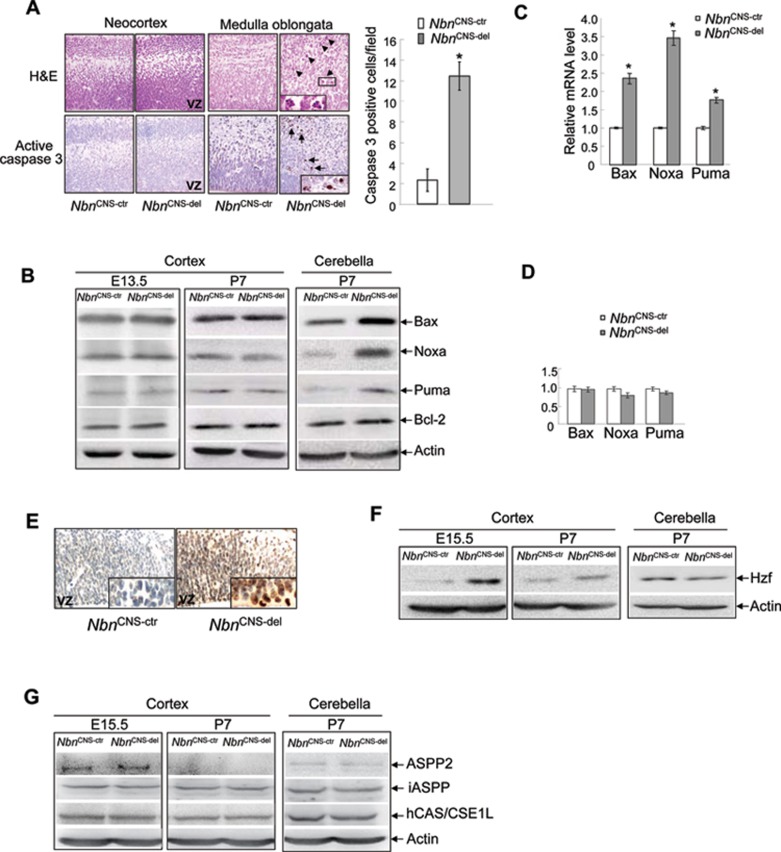
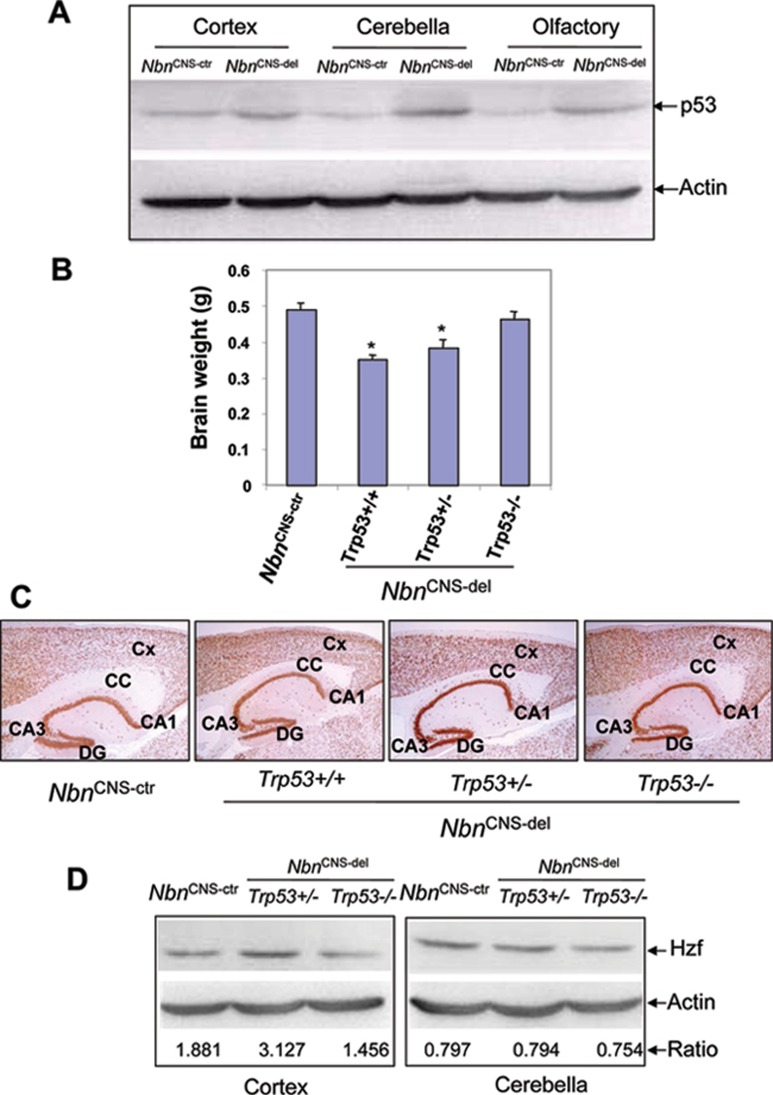
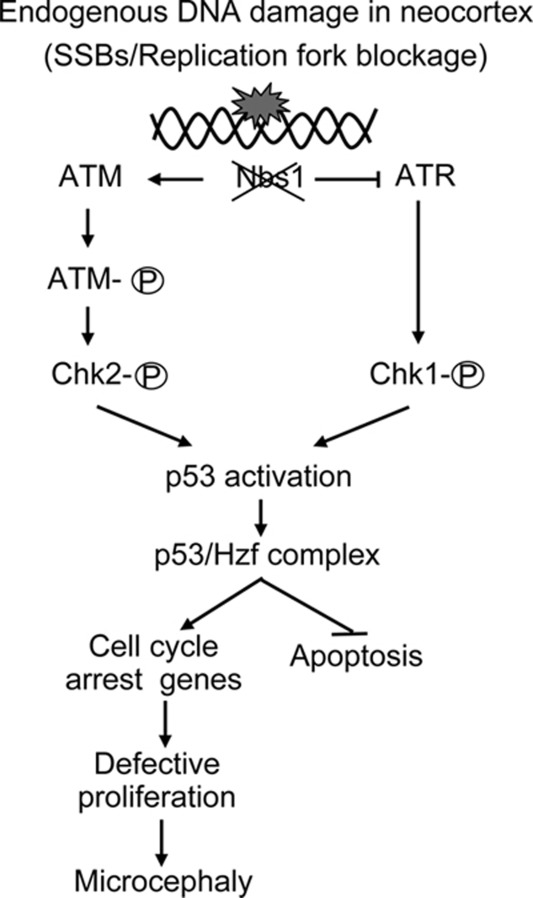
Microcephaly is a clinical characteristic for human nijmegen breakage syndrome (NBS, mutated in NBS1 gene), a chromosomal instability syndrome. However, the underlying molecular pathogenesis remains elusive. In the present study, we demonstrate that neuronal disruption of NBS (Nbn in mice) causes microcephaly characterized by the reduction of cerebral cortex and corpus callosum, recapitulating neuronal anomalies in human NBS. Nbs1-deficient neocortex shows accumulative endogenous DNA damage and defective activation of Ataxia telangiectasia and Rad3-related (ATR)-Chk1 pathway upon DNA damage. Notably, in contrast to massive apoptotic cell death in Nbs1-deficient cerebella, activation of p53 leads to a defective neuroprogenitor proliferation in neocortex, likely via specific persistent induction of hematopoietic zinc finger (Hzf) that preferentially promotes p53-mediated cell cycle arrest whilst inhibiting apoptosis. Moreover, Trp53 mutations substantially rescue the microcephaly in Nbs1-deficient mice. Thus, the present results reveal the first clue that developing neurons at different regions of brain selectively respond to endogenous DNA damage, and underscore an important role for Nbs1 in neurogenesis.
Figures
References
-
- Varon R, Vissinga C, Platzer M, et al. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell. 1998;93:467–476. - PubMed
-
- Riches LC, Lynch AM, Gooderham NJ. Early events in the mammalian response to DNA double-strand breaks. Mutagenesis. 2008;23:331–339. - PubMed
-
- Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308:551–554. - PubMed
-
- Difilippantonio S, Nussenzweig A. The NBS1-ATM connection revisited. Cell Cycle. 2007;6:2366–2370. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
