

Myeloperoxidase-derived oxidants inhibit sarco/endoplasmic reticulum Ca2+-ATPase activity and perturb Ca2+ homeostasis in human coronary artery endothelial cells
- PMID: 22214747
- PMCID: PMC3736816
- DOI: 10.1016/j.freeradbiomed.2011.12.001
Myeloperoxidase-derived oxidants inhibit sarco/endoplasmic reticulum Ca2+-ATPase activity and perturb Ca2+ homeostasis in human coronary artery endothelial cells
Abstract
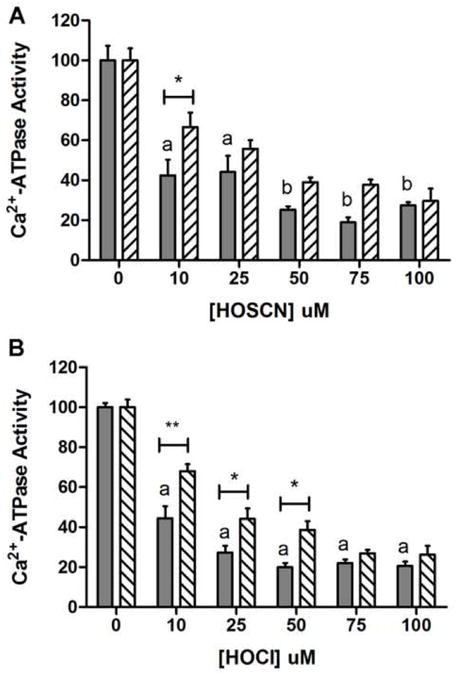
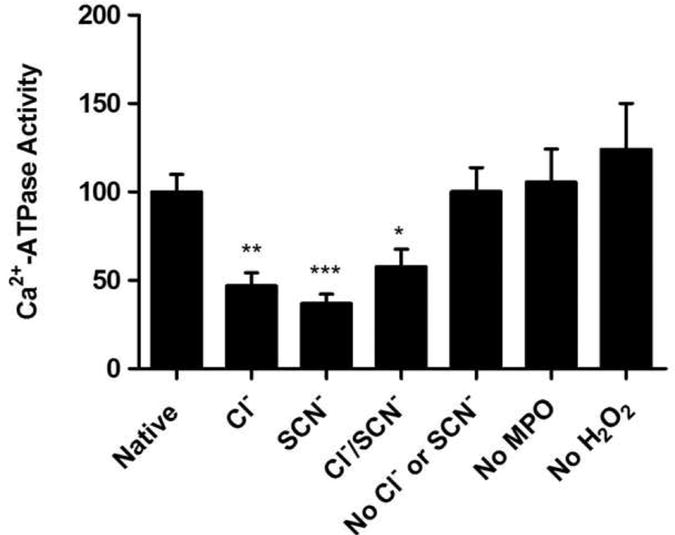
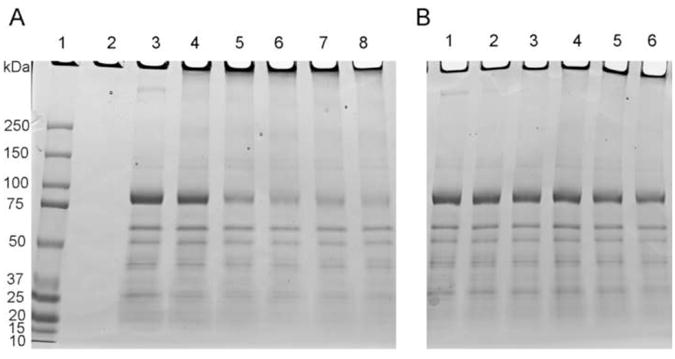
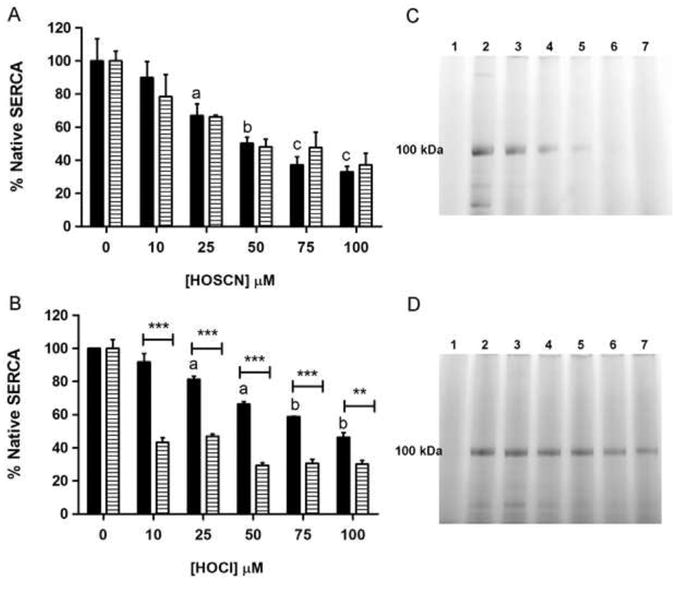
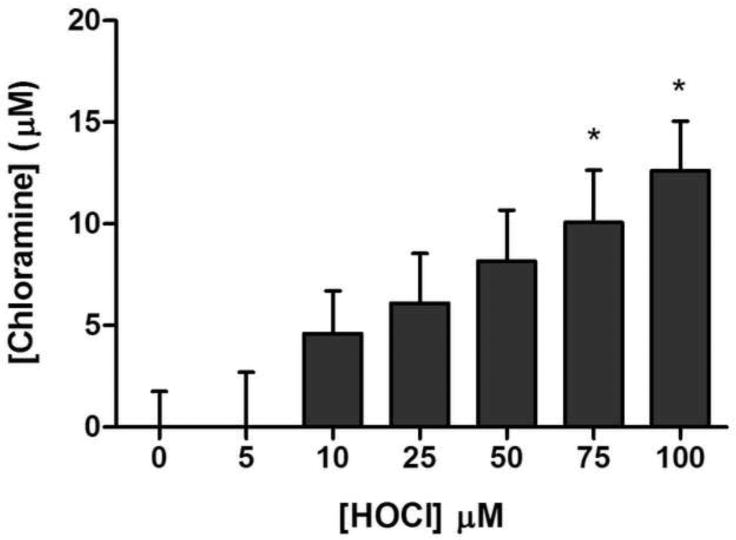
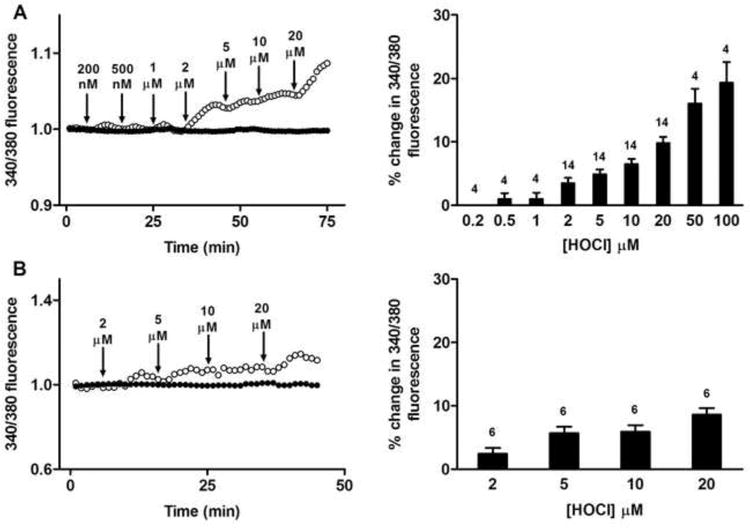
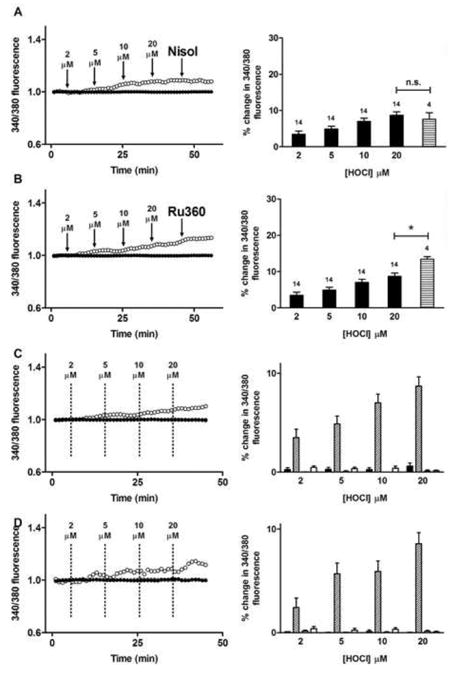
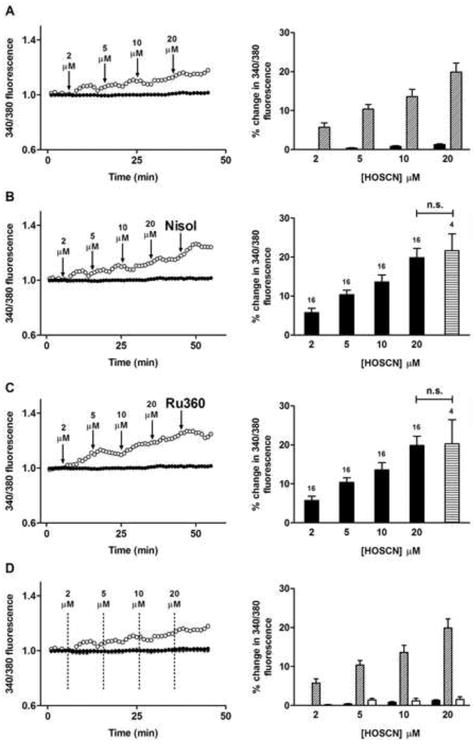
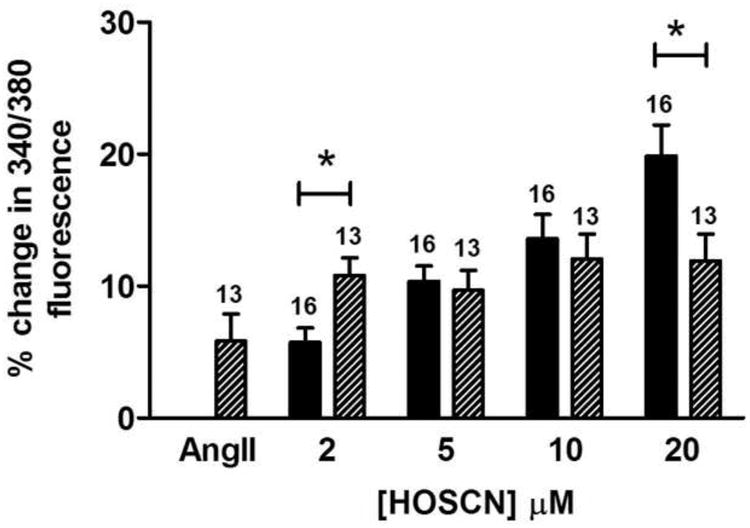
The sarco/endoplasmic reticulum Ca(2+)-ATPase (SERCA) plays a critical role in Ca(2+) homeostasis via sequestration of this ion in the sarco/endoplasmic reticulum. The activity of this pump is inhibited by oxidants and impaired in aging tissues and cardiovascular disease. We have shown previously that the myeloperoxidase (MPO)-derived oxidants HOCl and HOSCN target thiols and mediate cellular dysfunction. As SERCA contains Cys residues critical to ATPase activity, we hypothesized that HOCl and HOSCN might inhibit SERCA activity, via thiol oxidation, and increase cytosolic Ca(2+) levels in human coronary artery endothelial cells (HCAEC). Exposure of sarcoplasmic reticulum vesicles to preformed or enzymatically generated HOCl and HOSCN resulted in a concentration-dependent decrease in ATPase activity; this was also inhibited by the SERCA inhibitor thapsigargin. Decomposed HOSCN and incomplete MPO enzyme systems did not decrease activity. Loss of ATPase activity occurred concurrent with oxidation of SERCA Cys residues and protein modification. Exposure of HCAEC, with or without external Ca(2+), to HOSCN or HOCl resulted in a time- and concentration-dependent increase in intracellular Ca(2+) under conditions that did not result in immediate loss of cell viability. Thapsigargin, but not inhibitors of plasma membrane or mitochondrial Ca(2+) pumps/channels, completely attenuated the increase in intracellular Ca(2+) consistent with a critical role for SERCA in maintaining endothelial cell Ca(2+) homeostasis. Angiotensin II pretreatment potentiated the effect of HOSCN at low concentrations. MPO-mediated modulation of intracellular Ca(2+) levels may exacerbate endothelial dysfunction, a key early event in atherosclerosis, and be more marked in smokers because of their higher SCN(-) levels.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Comparative reactivity of the myeloperoxidase-derived oxidants hypochlorous acid and hypothiocyanous acid with human coronary artery endothelial cells.Free Radic Biol Med. 2013 Dec;65:1352-1362. doi: 10.1016/j.freeradbiomed.2013.10.007. Epub 2013 Oct 10. Free Radic Biol Med. 2013. PMID: 24120969
-
Low-density lipoprotein modified by myeloperoxidase oxidants induces endothelial dysfunction.Redox Biol. 2017 Oct;13:623-632. doi: 10.1016/j.redox.2017.08.004. Epub 2017 Aug 5. Redox Biol. 2017. PMID: 28818791 Free PMC article.
-
Comparative reactivity of the myeloperoxidase-derived oxidants HOCl and HOSCN with low-density lipoprotein (LDL): Implications for foam cell formation in atherosclerosis.Arch Biochem Biophys. 2015 May 1;573:40-51. doi: 10.1016/j.abb.2015.03.008. Epub 2015 Mar 18. Arch Biochem Biophys. 2015. PMID: 25795019
-
Reactions and reactivity of myeloperoxidase-derived oxidants: differential biological effects of hypochlorous and hypothiocyanous acids.Free Radic Res. 2012 Aug;46(8):975-95. doi: 10.3109/10715762.2012.667566. Epub 2012 Apr 23. Free Radic Res. 2012. PMID: 22348603 Review.
-
Sarco-Endoplasmic Reticulum Calcium Release Model Based on Changes in the Luminal Calcium Content.Adv Exp Med Biol. 2020;1131:337-370. doi: 10.1007/978-3-030-12457-1_14. Adv Exp Med Biol. 2020. PMID: 31646517 Review.
Cited by
-
Serum calcium levels are not associated with coronary heart disease.Vasc Health Risk Manag. 2013;9:517-20. doi: 10.2147/VHRM.S49136. Epub 2013 Sep 3. Vasc Health Risk Manag. 2013. PMID: 24043943 Free PMC article.
-
Mechanistic Role of the Calcium-Dependent Protease Calpain in the Endothelial Dysfunction Induced by MPO (Myeloperoxidase).Hypertension. 2018 Apr;71(4):761-770. doi: 10.1161/HYPERTENSIONAHA.117.10305. Hypertension. 2018. PMID: 29507101 Free PMC article.
-
Chlorinative stress in age-related diseases: a literature review.Immun Ageing. 2017 Nov 14;14:21. doi: 10.1186/s12979-017-0104-5. eCollection 2017. Immun Ageing. 2017. PMID: 29163665 Free PMC article. Review.
-
Myeloperoxidase evokes substantial vasomotor responses in isolated skeletal muscle arterioles of the rat.Acta Physiol (Oxf). 2015 May;214(1):109-23. doi: 10.1111/apha.12488. Epub 2015 Mar 28. Acta Physiol (Oxf). 2015. PMID: 25760778 Free PMC article.
-
Fibrosis in Atrial Fibrillation - Role of Reactive Species and MPO.Front Physiol. 2012 Jun 20;3:214. doi: 10.3389/fphys.2012.00214. eCollection 2012. Front Physiol. 2012. PMID: 22723783 Free PMC article.
References
-
- Davies MJ, Hawkins CL, Pattison DI, Rees MD. Mammalian heme peroxidases: from molecular mechanisms to health implications. Antioxid Redox Signal. 2008;10:1199–1234. - PubMed
-
- van der Veen BS, de Winther MP, Heeringa P. Myeloperoxidase: molecular mechanisms of action and their relevance to human health and disease. Antioxid Redox Signal. 2009;11:2899–2937. - PubMed
-
- Zhang R, Brennan ML, Fu X, Aviles RJ, Pearce GL, Penn MS, Topol EJ, Sprecher DL, Hazen SL. Association between myeloperoxidase levels and risk of coronary artery disease. JAMA. 2001;286:2136–2142. - PubMed
-
- Brennan ML, Penn MS, Van Lente F, Nambi V, Shishehbor MH, Aviles RJ, Goormastic M, Pepoy ML, McErlean ES, Topol EJ, Nissen SE, Hazen SL. Prognostic value of myeloperoxidase in patients with chest pain. N Engl J Med. 2003;349:1595–1604. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
