

Serum amyloid A opposes lipoxin A₄ to mediate glucocorticoid refractory lung inflammation in chronic obstructive pulmonary disease
- PMID: 22215599
- PMCID: PMC3271884
- DOI: 10.1073/pnas.1109382109
Serum amyloid A opposes lipoxin A₄ to mediate glucocorticoid refractory lung inflammation in chronic obstructive pulmonary disease
Abstract
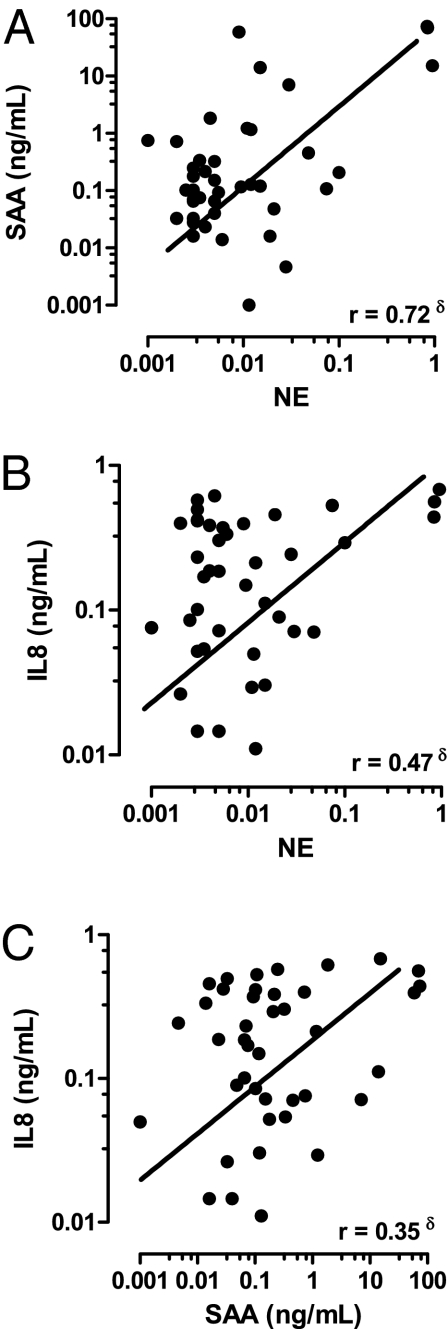
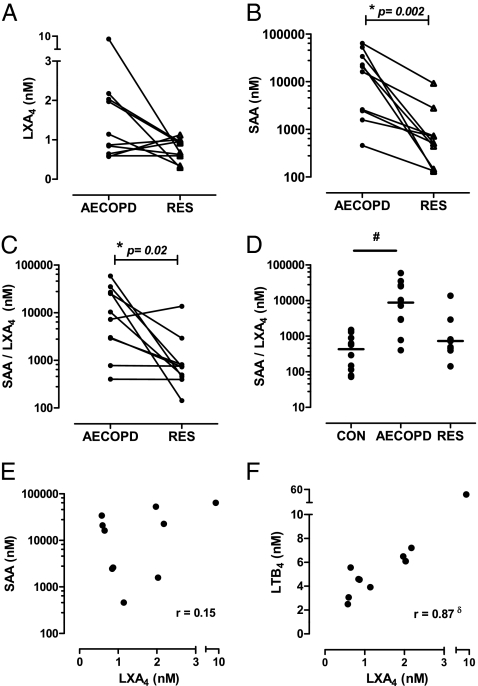
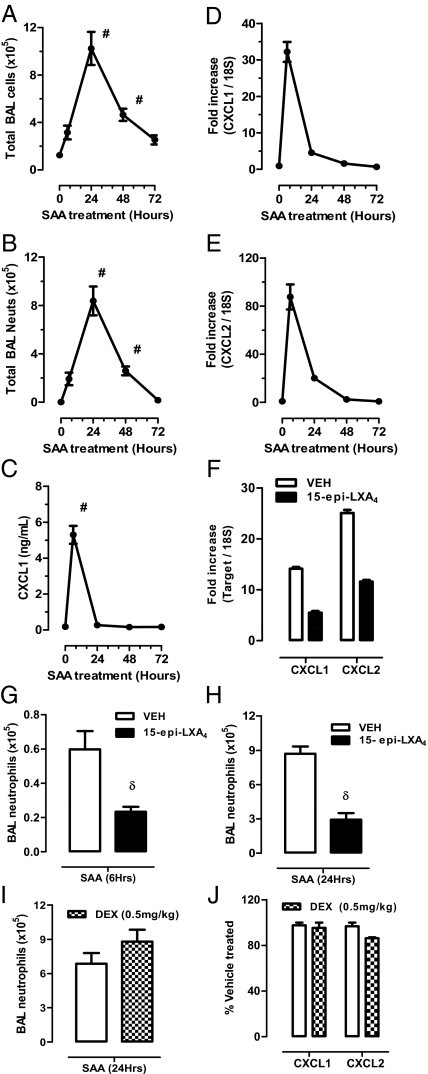
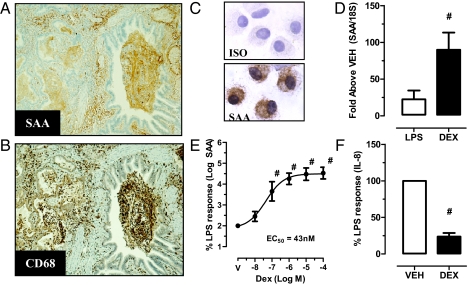
Chronic obstructive pulmonary disease (COPD) will soon be the third most common cause of death globally. Despite smoking cessation, neutrophilic mucosal inflammation persistently damages the airways and fails to protect from recurrent infections. This maladaptive and excess inflammation is also refractory to glucocorticosteroids (GC). Here, we identify serum amyloid A (SAA) as a candidate mediator of GC refractory inflammation in COPD. Extrahepatic SAA was detected locally in COPD bronchoalveolar lavage fluid, which correlated with IL-8 and neutrophil elastase, consistent with neutrophil recruitment and activation. Immunohistochemistry detected SAA was in close proximity to airway epithelium, and in vitro SAA triggered release of IL-8 and other proinflammatory mediators by airway epithelial cells in an ALX/FPR2 (formyl peptide receptor 2) receptor-dependent manner. Lipoxin A(4) (LXA(4)) can also interact with ALX/FPR2 receptors and lead to allosteric inhibition of SAA-initiated epithelial responses (pA(2) 13 nM). During acute exacerbation, peripheral blood SAA levels increased dramatically and were disproportionately increased relative to LXA(4). Human lung macrophages (CD68(+)) colocalized with SAA and GCs markedly increased SAA in vitro (THP-1, pEC(50) 43 nM). To determine its direct actions, SAA was administered into murine lung, leading to induction of CXC chemokine ligand 1/2 and a neutrophilic response that was inhibited by 15-epi-LXA(4) but not dexamethasone. Taken together, these findings identify SAA as a therapeutic target for inhibition and implicate SAA as a mediator of GC-resistant lung inflammation that can overwhelm organ protective signaling by lipoxins at ALX/FPR2 receptors.
Conflict of interest statement
Conflict of interest statement: B.D.L. is a co-inventor on patents on lipoxins in airway disease that are assigned to Brigham and Women's Hospital and licensed for clinical development.
Figures

Similar articles
-
Treating neutrophilic inflammation in COPD by targeting ALX/FPR2 resolution pathways.Pharmacol Ther. 2013 Dec;140(3):280-9. doi: 10.1016/j.pharmthera.2013.07.007. Epub 2013 Jul 21. Pharmacol Ther. 2013. PMID: 23880288 Review.
-
ALX/FPR2 Contributes to Serum Amyloid A-Induced Lung Neutrophil Recruitment Following Acute Ozone Exposure.FASEB J. 2025 Jun 15;39(11):e70555. doi: 10.1096/fj.202402865R. FASEB J. 2025. PMID: 40420730 Free PMC article.
-
Lack of activity of 15-epi-lipoxin A₄ on FPR2/ALX and CysLT1 receptors in interleukin-8-driven human neutrophil function.Clin Exp Immunol. 2013 Aug;173(2):298-309. doi: 10.1111/cei.12110. Clin Exp Immunol. 2013. PMID: 23607720 Free PMC article.
-
Serum amyloid A promotes lung neutrophilia by increasing IL-17A levels in the mucosa and γδ T cells.Am J Respir Crit Care Med. 2013 Jul 15;188(2):179-86. doi: 10.1164/rccm.201211-2139OC. Am J Respir Crit Care Med. 2013. PMID: 23627303 Free PMC article.
-
Opposing regulation of neutrophil apoptosis through the formyl peptide receptor-like 1/lipoxin A4 receptor: implications for resolution of inflammation.J Leukoc Biol. 2008 Sep;84(3):600-6. doi: 10.1189/jlb.1107765. Epub 2008 May 21. J Leukoc Biol. 2008. PMID: 18495783 Review.
Cited by
-
COPD and squamous cell lung cancer: aberrant inflammation and immunity is the common link.Br J Pharmacol. 2016 Feb;173(4):635-48. doi: 10.1111/bph.13198. Epub 2015 Jul 8. Br J Pharmacol. 2016. PMID: 26013585 Free PMC article. Review.
-
An investigation of the resolution of inflammation (catabasis) in COPD.Respir Res. 2012 Nov 13;13(1):101. doi: 10.1186/1465-9921-13-101. Respir Res. 2012. PMID: 23148928 Free PMC article.
-
Serum Amyloid A Contributes to Chronic Apical Periodontitis via TLR2 and TLR4.J Dent Res. 2019 Jan;98(1):117-125. doi: 10.1177/0022034518796456. Epub 2018 Sep 6. J Dent Res. 2019. PMID: 30189157 Free PMC article.
-
Effects of Smoking on Inflammatory-Related Cytokine Levels in Human Serum.Molecules. 2022 Jun 9;27(12):3715. doi: 10.3390/molecules27123715. Molecules. 2022. PMID: 35744838 Free PMC article.
-
Serum amyloid A, protein Z, and C4b-binding protein β chain as new potential biomarkers for pulmonary tuberculosis.PLoS One. 2017 Mar 9;12(3):e0173304. doi: 10.1371/journal.pone.0173304. eCollection 2017. PLoS One. 2017. PMID: 28278182 Free PMC article.
References
-
- Barnes PJ, Adcock IM. Glucocorticoid resistance in inflammatory diseases. Lancet. 2009;373:1905–1917. - PubMed
-
- Willemse BW, et al. Effect of 1-year smoking cessation on airway inflammation in COPD and asymptomatic smokers. Eur Respir J. 2005;26:835–845. - PubMed
-
- Pauwels RA, Rabe KF. Burden and clinical features of chronic obstructive pulmonary disease (COPD) Lancet. 2004;364:613–620. - PubMed
-
- Serhan CN, Savill J. Resolution of inflammation: The beginning programs the end. Nat Immunol. 2005;6:1191–1197. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
