

A decade of 3C technologies: insights into nuclear organization
- PMID: 22215806
- PMCID: PMC3258961
- DOI: 10.1101/gad.179804.111
A decade of 3C technologies: insights into nuclear organization
Abstract
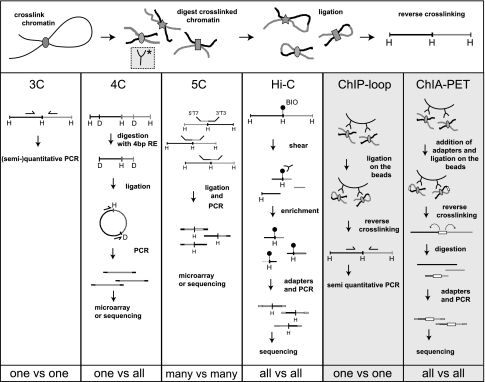
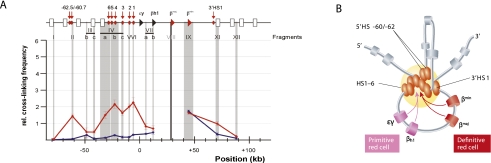
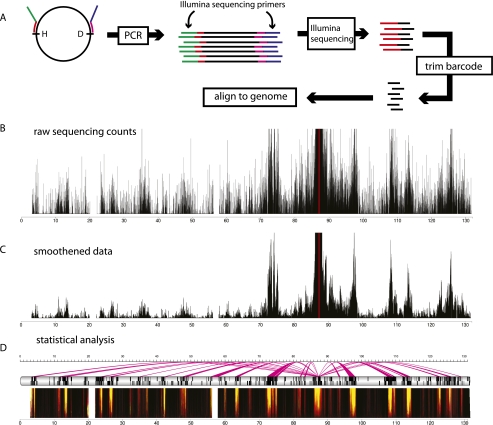
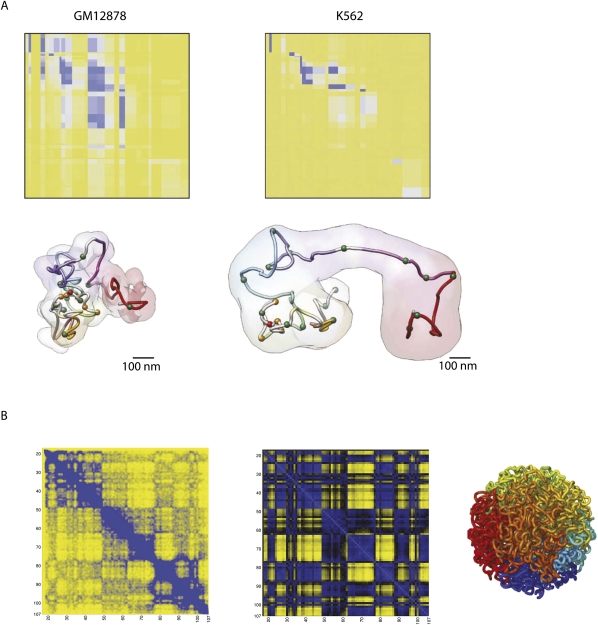
Over the past 10 years, the development of chromosome conformation capture (3C) technology and the subsequent genomic variants thereof have enabled the analysis of nuclear organization at an unprecedented resolution and throughput. The technology relies on the original and, in hindsight, remarkably simple idea that digestion and religation of fixed chromatin in cells, followed by the quantification of ligation junctions, allows for the determination of DNA contact frequencies and insight into chromosome topology. Here we evaluate and compare the current 3C-based methods (including 4C [chromosome conformation capture-on-chip], 5C [chromosome conformation capture carbon copy], HiC, and ChIA-PET), summarize their contribution to our current understanding of genome structure, and discuss how shape influences genome function.
Figures
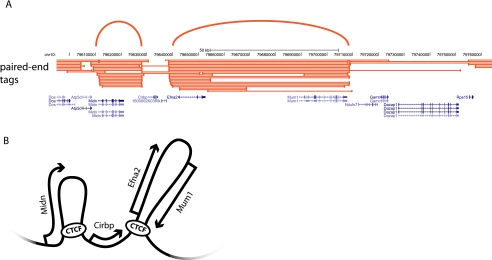
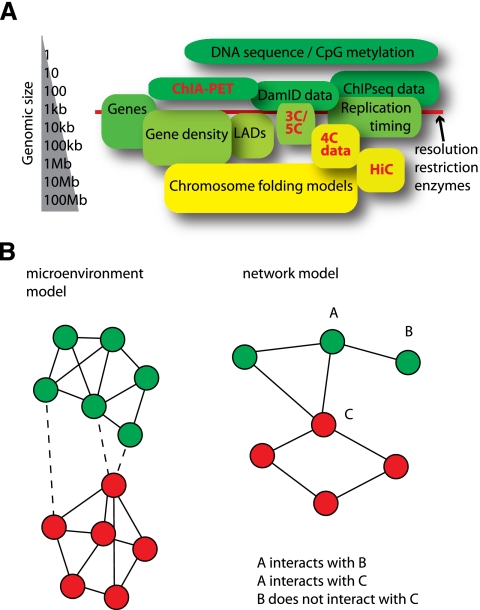
References
-
- Al-Shahrour F, Diaz-Uriarte R, Dopazo J 2004. FatiGO: A Web tool for finding significant associations of gene ontology terms with groups of genes. Bioinformatics 20: 578–580 - PubMed
-
- Bantignies F, Roure V, Comet I, Leblanc B, Schuettengruber B, Bonnet J, Tixier V, Mas A, Cavalli G 2011. Polycomb-dependent regulatory contacts between distant Hox loci in Drosophila. Cell 144: 214–226 - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous