

A crucial role of activin A-mediated growth hormone suppression in mouse and human heart failure
- PMID: 22216087
- PMCID: PMC3247209
- DOI: 10.1371/journal.pone.0027901
A crucial role of activin A-mediated growth hormone suppression in mouse and human heart failure
Abstract
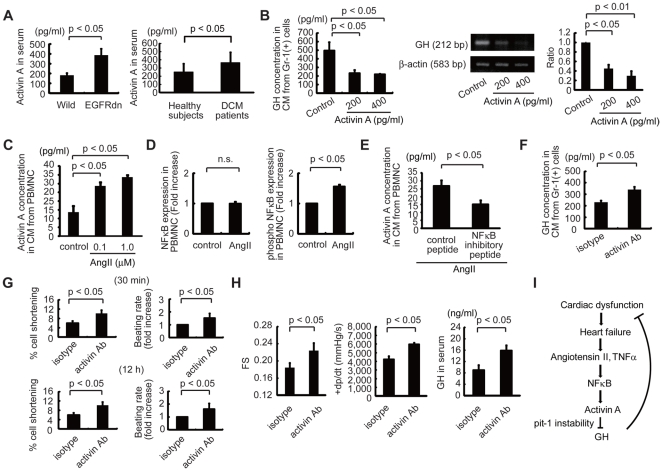
Infusion of bone marrow-derived mononuclear cells (BMMNC) has been reported to ameliorate cardiac dysfunction after acute myocardial infarction. In this study, we investigated whether infusion of BMMNC is also effective for non-ischemic heart failure model mice and the underlying mechanisms. Intravenous infusion of BMMNC showed transient cardioprotective effects on animal models with dilated cardiomyopathy (DCM) without their engraftment in heart, suggesting that BMMNC infusion improves cardiac function via humoral factors rather than their differentiation into cardiomyocytes. Using conditioned media from sorted BMMNC, we found that the cardioprotective effects were mediated by growth hormone (GH) secreted from myeloid (Gr-1(+)) cells and the effects was partially mediated by signal transducer and activator of transcription 3 in cardiomyocytes. On the other hand, the GH expression in Gr-1(+) cells was significantly downregulated in DCM mice compared with that in healthy control, suggesting that the environmental cue in heart failure might suppress the Gr-1(+) cells function. Activin A was upregulated in the serum of DCM models and induced downregulation of GH levels in Gr-1(+) cells and serum. Furthermore, humoral factors upregulated in heart failure including angiotensin II upregulated activin A in peripheral blood mononuclear cells (PBMNC) via activation of NFκB. Similarly, serum activin A levels were also significantly higher in DCM patients with heart failure than in healthy subjects and the GH levels in conditioned medium from PBMNC of DCM patients were lower than that in healthy subjects. Inhibition of activin A increased serum GH levels and improved cardiac function of DCM model mice. These results suggest that activin A causes heart failure by suppressing GH activity and that inhibition of activin A might become a novel strategy for the treatment of heart failure.
© 2011 Frank Emmert-Streib
Conflict of interest statement
Figures





Similar articles
-
Activin/follistatin system in grass carp pituitary cells: - Regulation by local release of growth hormone and luteinizing hormone and its functional role in growth hormone synthesis and secretion.PLoS One. 2017 Jun 29;12(6):e0179789. doi: 10.1371/journal.pone.0179789. eCollection 2017. PLoS One. 2017. PMID: 28662143 Free PMC article.
-
Recombinant human growth hormone treatment for dilated cardiomyopathy in children.Pediatrics. 2004 Oct;114(4):e452-8. doi: 10.1542/peds.2004-0072. Pediatrics. 2004. PMID: 15466071 Clinical Trial.
-
Activin-A modulates growth hormone secretion from cultures of rat anterior pituitary cells.Endocrinology. 1990 May;126(5):2369-76. doi: 10.1210/endo-126-5-2369. Endocrinology. 1990. PMID: 2158424
-
Role of growth hormone in chronic heart failure: therapeutic implications.Ital Heart J. 2000 Nov;1(11):732-8. Ital Heart J. 2000. PMID: 11110515 Review.
-
Growth hormone, acromegaly, and heart failure: an intricate triangulation.Clin Endocrinol (Oxf). 2003 Dec;59(6):660-71. doi: 10.1046/j.1365-2265.2003.01780.x. Clin Endocrinol (Oxf). 2003. PMID: 14974906 Review.
Cited by
-
Diminished vasculogenesis under inflammatory conditions is mediated by Activin A.Angiogenesis. 2023 Aug;26(3):423-436. doi: 10.1007/s10456-023-09873-w. Epub 2023 Mar 29. Angiogenesis. 2023. PMID: 36977946
-
Ramipril attenuates left ventricular remodeling by regulating the expression of activin A-follistatin in a rat model of heart failure.Sci Rep. 2016 Sep 19;6:33677. doi: 10.1038/srep33677. Sci Rep. 2016. PMID: 27642098 Free PMC article.
-
Activin A inhibition attenuates sympathetic neural remodeling following myocardial infarction in rats.Mol Med Rep. 2018 Apr;17(4):5074-5080. doi: 10.3892/mmr.2018.8496. Epub 2018 Jan 25. Mol Med Rep. 2018. PMID: 29393433 Free PMC article.
-
Sustained Elevated Circulating Activin A Impairs Global Longitudinal Strain in Pregnant Rats: A Potential Mechanism for Preeclampsia-Related Cardiac Dysfunction.Cells. 2022 Feb 21;11(4):742. doi: 10.3390/cells11040742. Cells. 2022. PMID: 35203391 Free PMC article.
-
Activin A Predicts Left Ventricular Remodeling and Mortality in Patients with ST-Elevation Myocardial Infarction.Acta Cardiol Sin. 2016 Jul;32(4):420-7. doi: 10.6515/acs20150415a. Acta Cardiol Sin. 2016. PMID: 27471355 Free PMC article.
References
-
- Passier R, van Laake LW, Mummery CL. Stem-cell-based therapy and lessons from the heart. Nature. 2008;453:322–329. - PubMed
-
- Schachinger V, Erbs S, Elsasser A, Haberbosch W, Hambrecht R, et al. Intracoronary bone marrow-derived progenitor cells in acute myocardial infarction. N Engl J Med. 2006;355:1210–1221. - PubMed
-
- Segers VF, Lee RT. Stem-cell therapy for cardiac disease. Nature. 2008;451:937–942. - PubMed
-
- van Ramshorst J, Bax JJ, Beeres SL, Dibbets-Schneider P, Roes SD, et al. Intramyocardial bone marrow cell injection for chronic myocardial ischemia: a randomized controlled trial. JAMA. 2009;301:1997–2004. - PubMed
-
- Martin-Rendon E, Brunskill SJ, Hyde CJ, Stanworth SJ, Mathur A, et al. Autologous bone marrow stem cells to treat acute myocardial infarction: a systematic review. Eur Heart J. 2008;29:1807–1818. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
