

doi: 10.1371/journal.pone.0028879.
Epub 2011 Dec 21.
A viral discovery methodology for clinical biopsy samples utilising massively parallel next generation sequencing
Affiliations
- PMID: 22216131
- PMCID: PMC3244418
- DOI: 10.1371/journal.pone.0028879
Item in Clipboard
A viral discovery methodology for clinical biopsy samples utilising massively parallel next generation sequencing
PLoS One.
2011.
Abstract
Here we describe a virus discovery protocol for a range of different virus genera, that can be applied to biopsy-sized tissue samples. Our viral enrichment procedure, validated using canine and human liver samples, significantly improves viral read copy number and increases the length of viral contigs that can be generated by de novo assembly. This in turn enables the Illumina next generation sequencing (NGS) platform to be used as an effective tool for viral discovery from tissue samples.
© 2011 Daly et al.
Conflict of interest statement
Figures
Extracted and pooled (×6) HCV infected biopsy RNA was reverse transcribed and amplified prior to Illumina NGS and mapped to the HCV reference 3a.NZL. NC_0009824 (Los Alamos HCV database).

a: Illustration of the key steps in isolating and enriching viral nucleic acids relative to host nucleic acids prior to sequencing by NGS. Liver tissue cells were broken in an isotonic buffer supplemented with BSA using a hard tissue probe and an Omni-Homogeniser with a dry-ice freeze/thaw step (repeated 3×). To ensure that cells membranes were broken the lysate was checked microscopically. b: Comparison of Alumina/pestle-grinding against hard-tissue probe homogenisation with freeze/thaw cycles (+/− the addition of Benzonase). (2 mm3 fragments were dissected from an HCV infected liver sample. Duplicate tissue samples were used for each protocol. HCV nucleic acids were measured in triplicate by dual labelled qPCR assay with NIBSC standard.
A SYBR-green assay and plasmid standard was used for the CAV and CPV samples, whilst the HCV and HBV assays used dual labelled probed with NIBSC standards. Total RNA extracted for HCV and Total DNA extracted for HBV, CPV and CAV. The Genomic strand copy number for HCV was estimated by performing the RT step in the presence of the reverse primer only.
For each viral liver sample, 2 mm3 of liver was used separately for total RNA extraction, total DNA extraction, DNA viral enrichment and RNA viral enrichment. qPCR assays were performed using 20 ng of each sample (estimated by Pico-green assay) as an input in triplicate. The samples subsequently mass sequenced are indicated with an asterisk.

To assess the relative difference between total RNA and total DNA extracts with the virally enriched samples, the NGS viral reads were mapped to respective reference genomes.
Viral reference sequencing reads shown as a percentage of total total reads from the Illumina NGS datasets for the differentially processed liver samples.
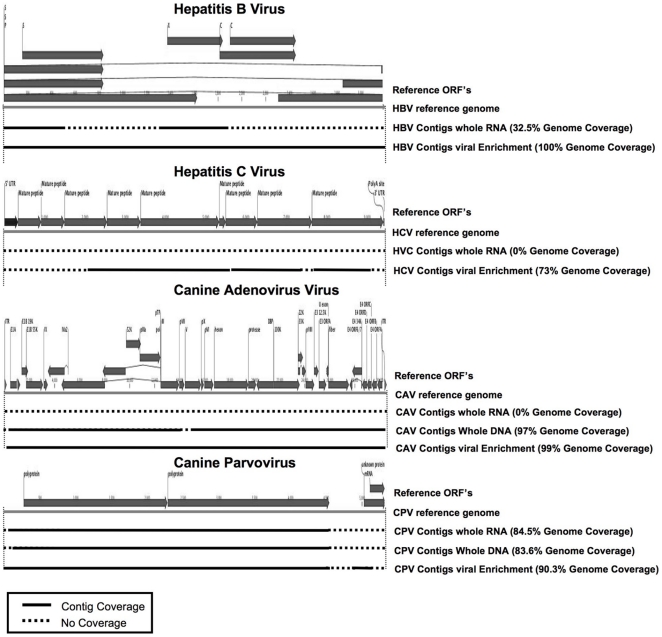
NGS reads for infected liver samples and their processed fractions, were used to generate contigs using ABySS and CLC de novo assembly algorithms. Contigs (length over 200 nt) were mapped to the viral reference genomes with the length and coverage indicated (solid black lines). Regions of the reference genomes not covered by the contigs are indicated with dashed lines.
References
-
- Reyes GR, Kim J. Sequence-independent, single-primer amplification (SISPA) of complex DNA populations. Mol Cell Probes. 1991;5:473–481. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
