

Implications of protein post-translational modifications in IBD
- PMID: 22223542
- PMCID: PMC3378042
- DOI: 10.1002/ibd.22859
Implications of protein post-translational modifications in IBD
Abstract
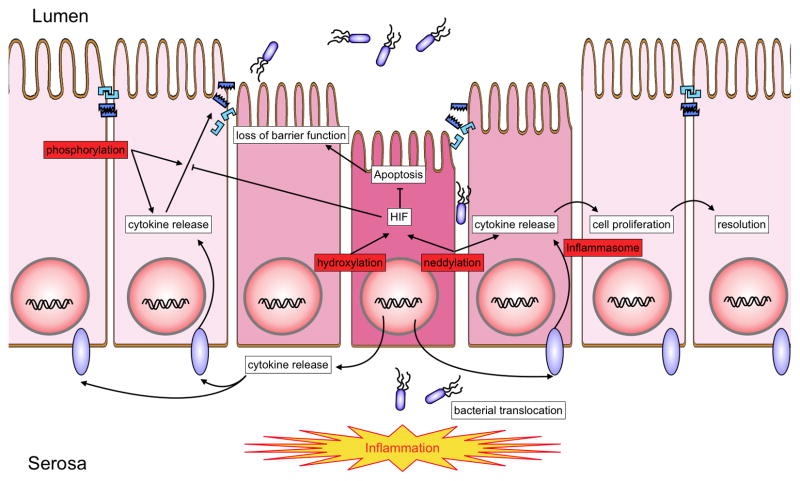
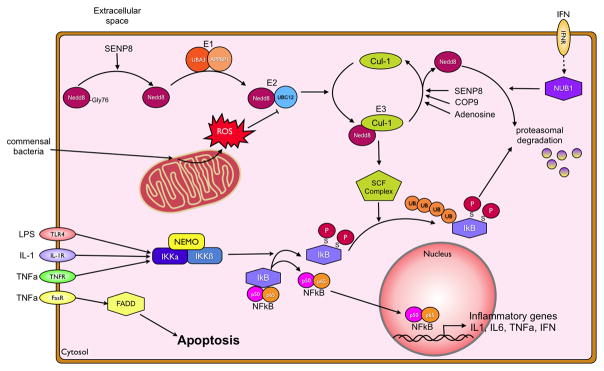
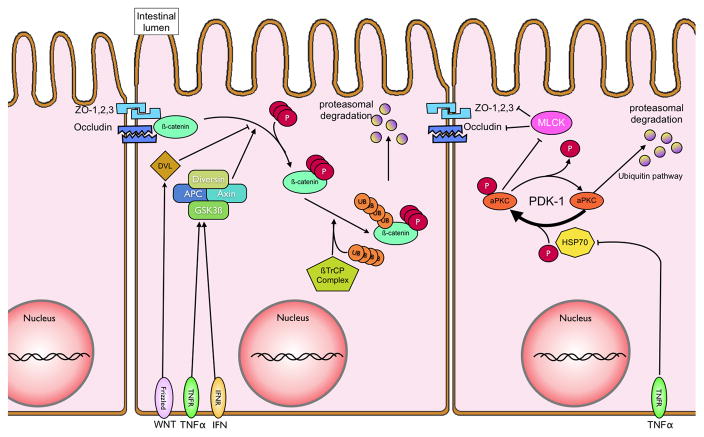
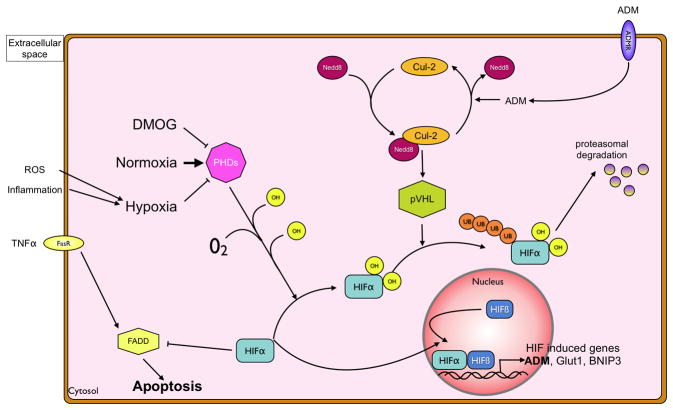
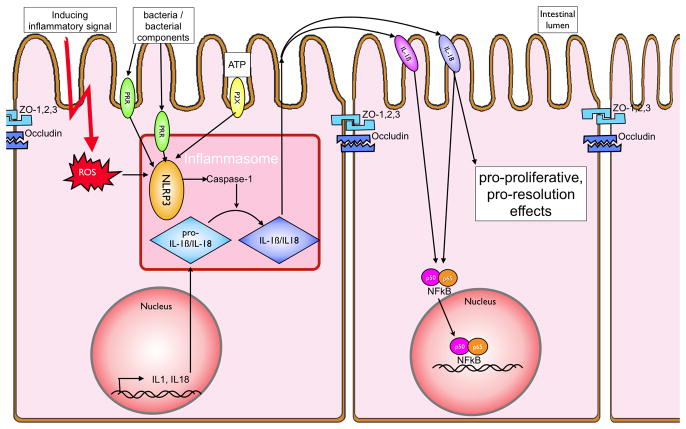
In recent years our understanding of the pathogenesis of inflammatory bowel disease (IBD) has greatly increased. Hallmarks of IBD include loss of intestinal barrier function, increased cytokine production, and failed resolution of tissue damage. Lasting treatments are still lacking and, therefore, a better understanding of the underlying molecular mechanisms is necessary to design novel therapeutic approaches. Apart from transcriptional and posttranscriptional regulation of relevant genes, mammals have evolved a complex and efficient series of mechanisms to rapidly modify newly made proteins for the purposes of signaling and adaptation. These posttranslational protein modifications include, among others, phosphorylation, hydroxylation, neddylation, and cytokine cleavage by the inflammasome. This review focuses on our current understanding of posttranslational protein modifications with a particular focus on their relevance to IBD pathogenesis.
Copyright © 2012 Crohn's & Colitis Foundation of America, Inc.
Figures
References
-
- Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. - PubMed
-
- Barnes PJ, Karin M. Nuclear factor-κB - A pivitol transcription factor in chronic inflammatory disease. N Engl J Med. 1997;336:1066–1071. - PubMed
-
- Wojcik C, Di Napoli M. Ubiquitin-proteasome system and proteasome inhibition: new strategies in stroke therapy. Stroke. 2004;35:1506–1518. Epub 2004 Apr 1529. - PubMed
-
- Chen Z, Hagler J, Palombella VJ, et al. Signal-induced site-specific phosphorylation targets I kappa B alpha to the ubiquitin-proteasome pathway. Genes Dev. 1995;9:1586–1597. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources