

Induction of DNA damage signaling upon Rift Valley fever virus infection results in cell cycle arrest and increased viral replication
- PMID: 22223653
- PMCID: PMC3293538
- DOI: 10.1074/jbc.M111.296608
Induction of DNA damage signaling upon Rift Valley fever virus infection results in cell cycle arrest and increased viral replication
Abstract
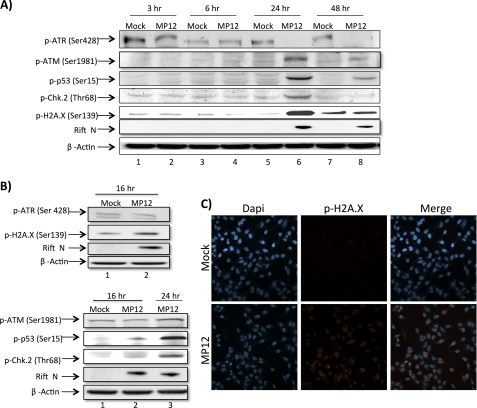
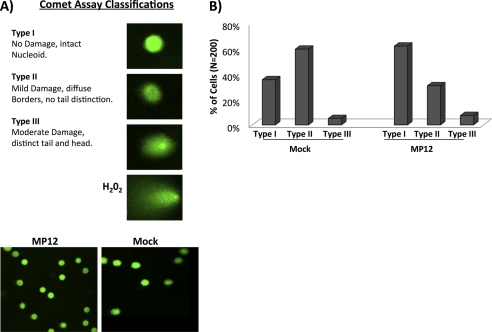
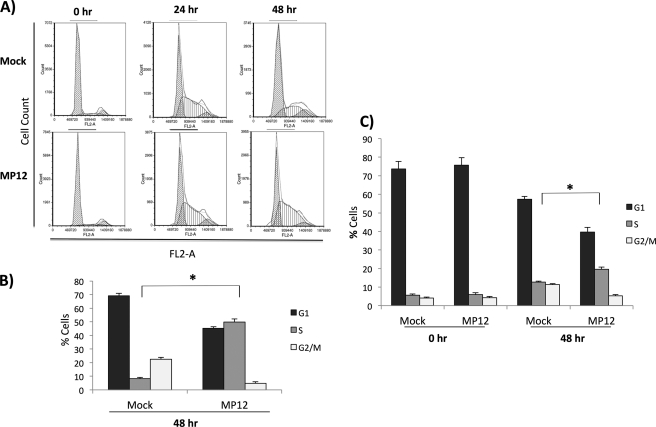
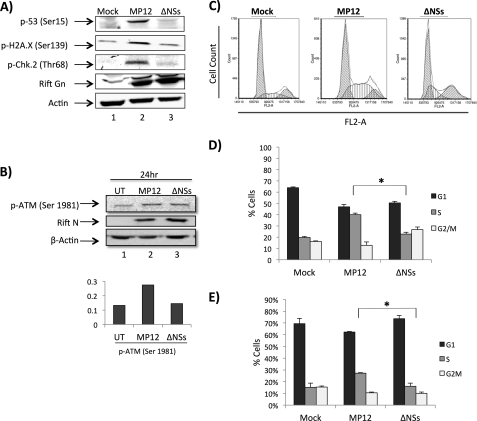
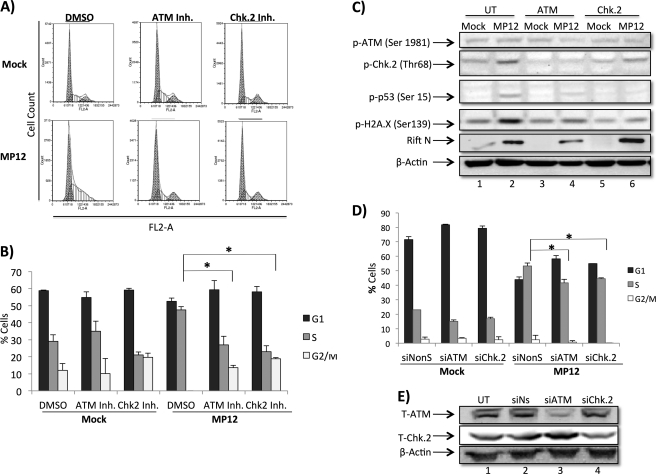
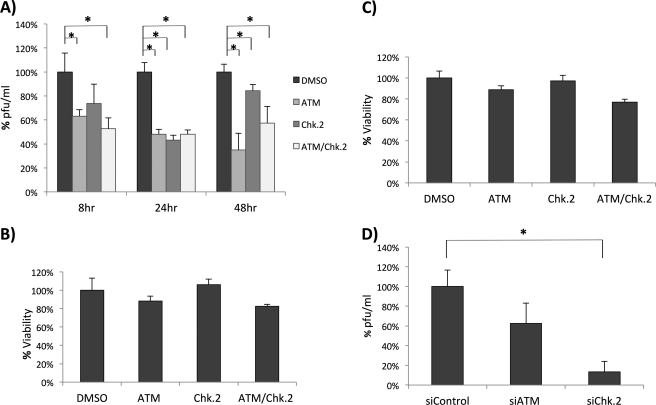
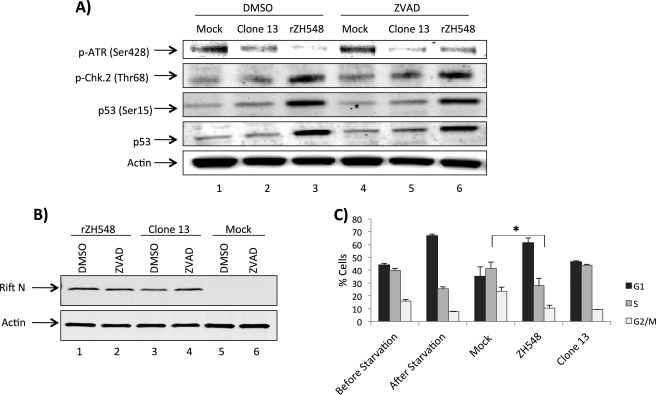
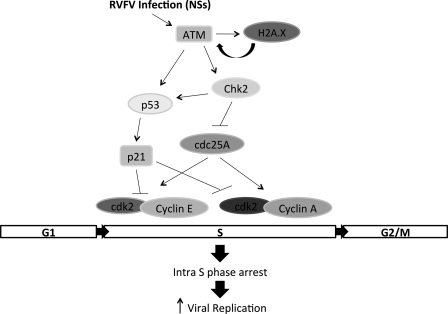
Rift Valley fever virus (RVFV) is a highly pathogenic arthropod-borne virus infecting a wide range of vertebrate hosts. Of particular interest is the nonstructural NSs protein, which forms large filamentous fibril bundles in the nucleus. Past studies have shown NSs to be a multifaceted protein important for virulence through modulation of the interferon response as well acting as a general inhibitor of transcription. Here we investigated the regulation of the DNA damage signaling cascades by RVFV infection and found virally inducted phosphorylation of the classical DNA damage signaling proteins, ataxia-telangiectasia mutated (ATM) (Ser-1981), Chk.2 (Thr-68), H2A.X (Ser-139), and p53 (Ser-15). In contrast, ataxia-telangiectasia mutated and Rad3-related kinase (ATR) (Ser-428) phosphorylation was decreased following RVFV infection. Importantly, both the attenuated vaccine strain MP12 and the fully virulent strain ZH548 showed strong parallels in their up-regulation of the ATM arm of the DNA damage response and in the down-regulation of the ATR pathway. The increase in DNA damage signaling proteins did not result from gross DNA damage as no increase in DNA damage was observed following infection. Rather the DNA damage signaling was found to be dependent on the viral protein NSs, as an NSs mutant virus was not found to induce the equivalent signaling pathways. RVFV MP12-infected cells also displayed an S phase arrest that was found to be dependent on NSs expression. Use of ATM and Chk.2 inhibitors resulted in a marked decrease in S phase arrest as well as viral production. These results indicate that RVFV NSs induces DNA damage signaling pathways that are beneficial for viral replication.
Figures
References
-
- Weber F., Elliott R. M. (2002) Antigenic drift, antigenic shift and interferon antagonists: how bunyaviruses counteract the immune system. Virus Res. 88, 129–136 - PubMed
-
- Bird B. H., Albariño C. G., Hartman A. L., Erickson B. R., Ksiazek T. G., Nichol S. T. (2008) Rift Valley fever virus lacking the NSs and NSm genes is highly attenuated, confers protective immunity from virulent virus challenge, and allows for differential identification of infected and vaccinated animals. J. Virol. 82, 2681–2691 - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
