

Mutations in mouse Ift144 model the craniofacial, limb and rib defects in skeletal ciliopathies
- PMID: 22228095
- PMCID: PMC3313797
- DOI: 10.1093/hmg/ddr613
Mutations in mouse Ift144 model the craniofacial, limb and rib defects in skeletal ciliopathies
Abstract
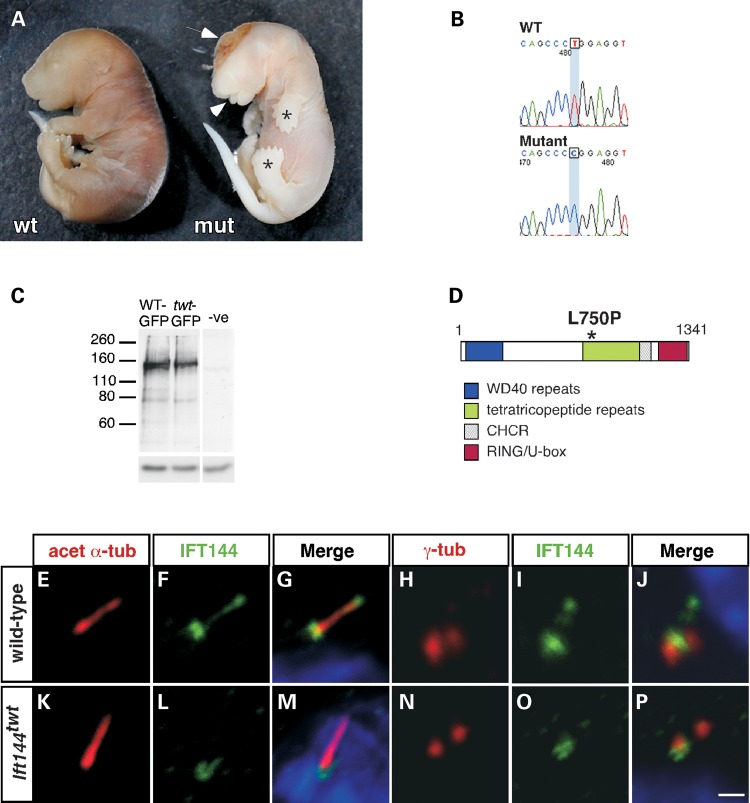
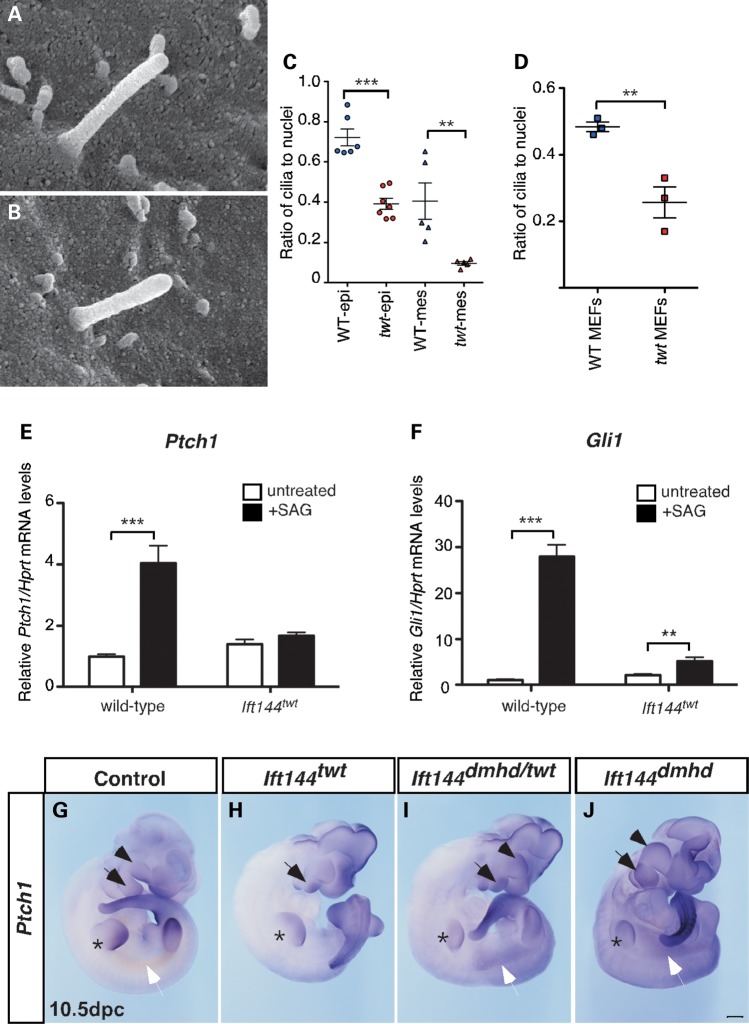
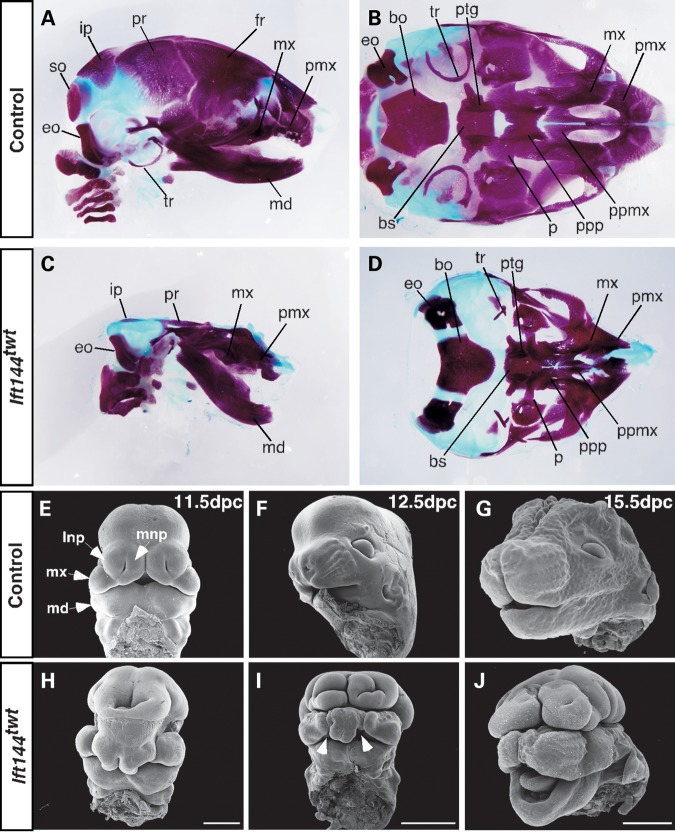
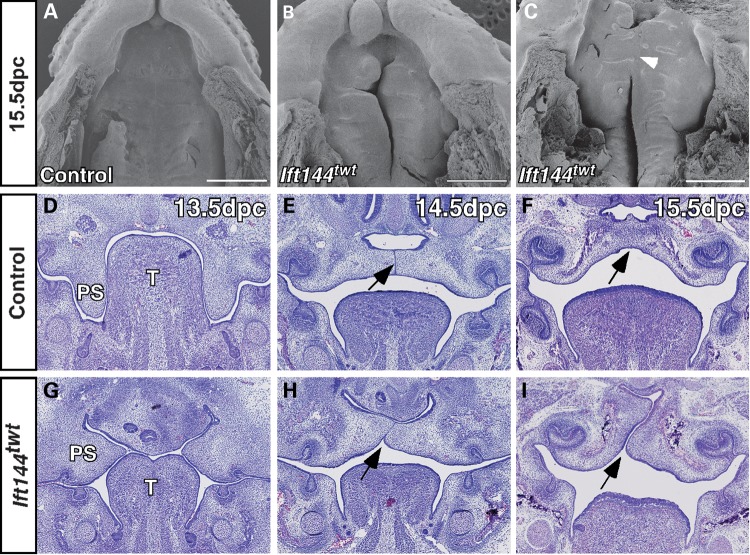
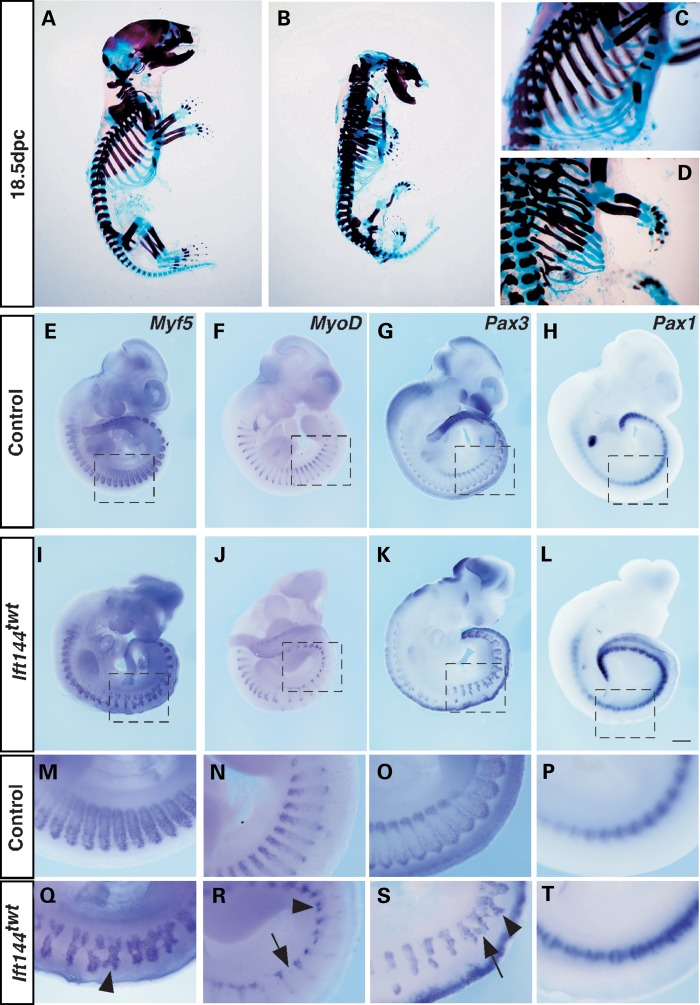
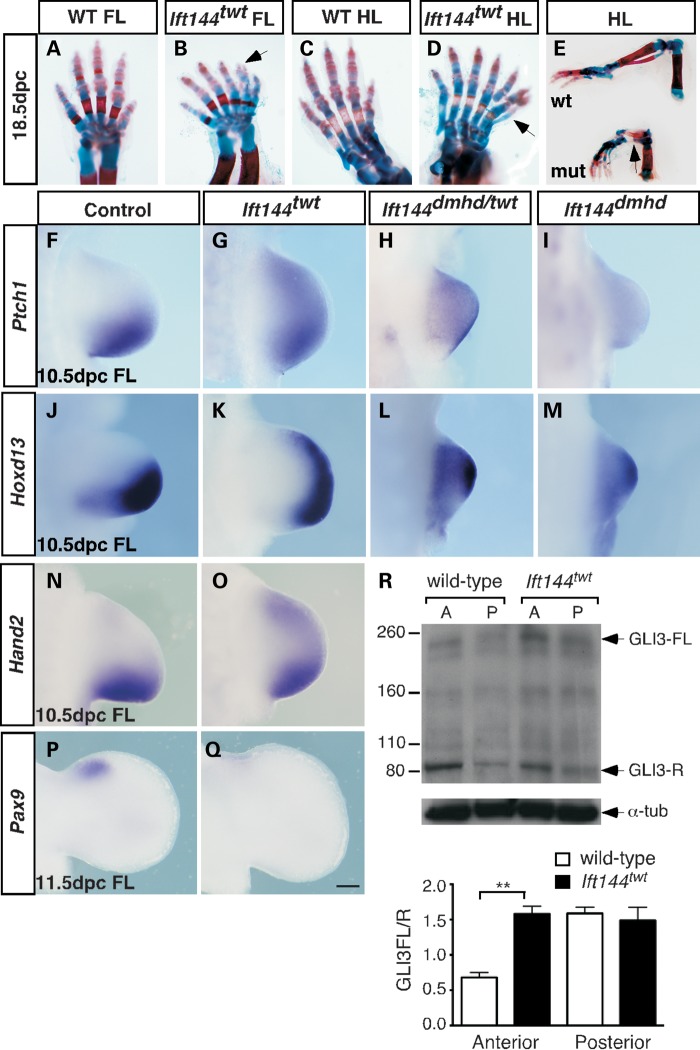
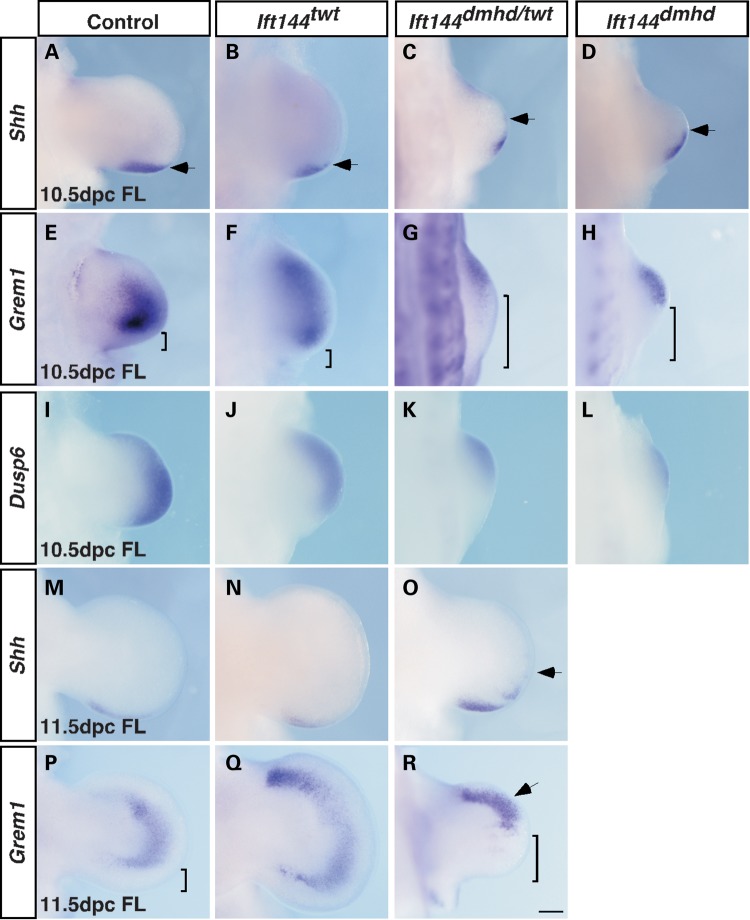
Mutations in components of the intraflagellar transport (IFT) machinery required for assembly and function of the primary cilium cause a subset of human ciliopathies characterized primarily by skeletal dysplasia. Recently, mutations in the IFT-A gene IFT144 have been described in patients with Sensenbrenner and Jeune syndromes, which are associated with short ribs and limbs, polydactyly and craniofacial defects. Here, we describe an N-ethyl-N-nitrosourea-derived mouse mutant with a hypomorphic missense mutation in the Ift144 gene. The mutant twinkle-toes (Ift144(twt)) phenocopies a number of the skeletal and craniofacial anomalies seen in patients with human skeletal ciliopathies. Like other IFT-A mouse mutants, Ift144 mutant embryos display a generalized ligand-independent expansion of hedgehog (Hh) signalling, in spite of defective ciliogenesis and an attenuation of the ability of mutant cells to respond to upstream stimulation of the pathway. This enhanced Hh signalling is consistent with cleft palate and polydactyly phenotypes in the Ift144(twt) mutant, although extensive rib branching, fusion and truncation phenotypes correlate with defects in early somite patterning and may reflect contributions from multiple signalling pathways. Analysis of embryos harbouring a second allele of Ift144 which represents a functional null, revealed a dose-dependent effect on limb outgrowth consistent with the short-limb phenotypes characteristic of these ciliopathies. This allelic series of mouse mutants provides a unique opportunity to uncover the underlying mechanistic basis of this intriguing subset of ciliopathies.
Figures
References
-
- Beales P.L., Bland E., Tobin J.L., Bacchelli C., Tuysuz B., Hill J., Rix S., Pearson C.G., Kai M., Hartley J., et al. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat. Genet. 2007;39:727–729. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
