

LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1
- PMID: 22228096
- PMCID: PMC3315202
- DOI: 10.1093/hmg/dds003
LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1
Abstract
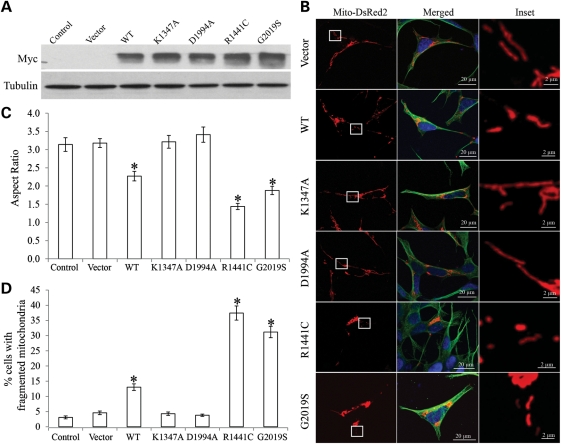
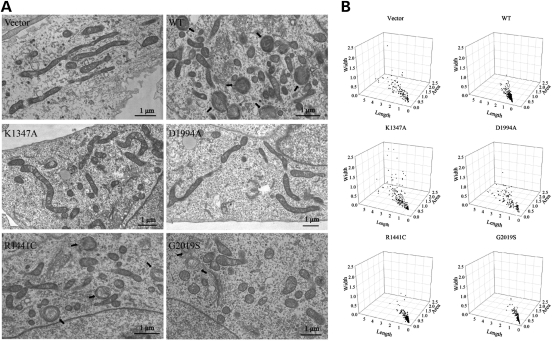
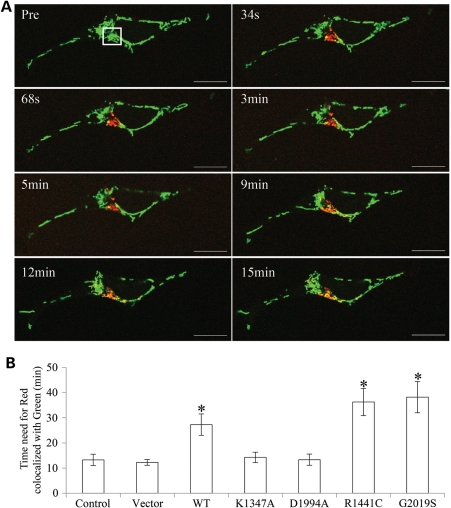
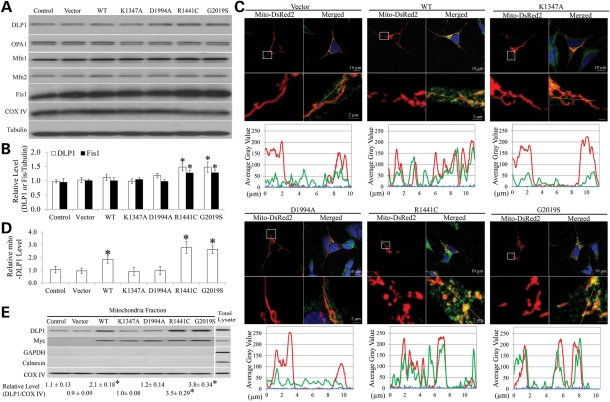
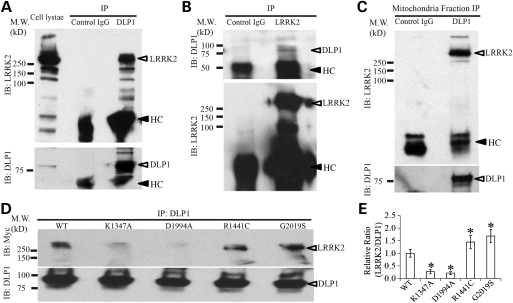
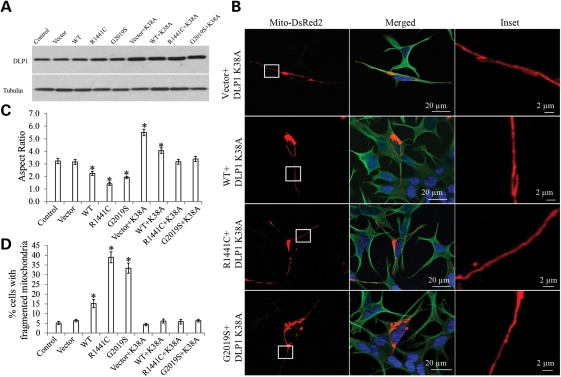
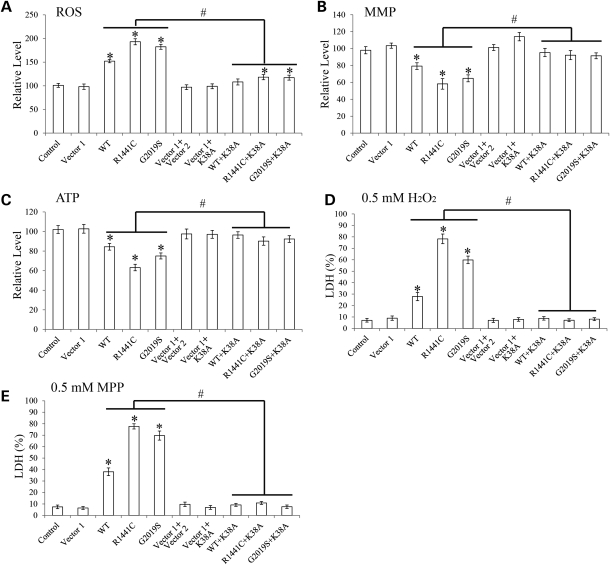
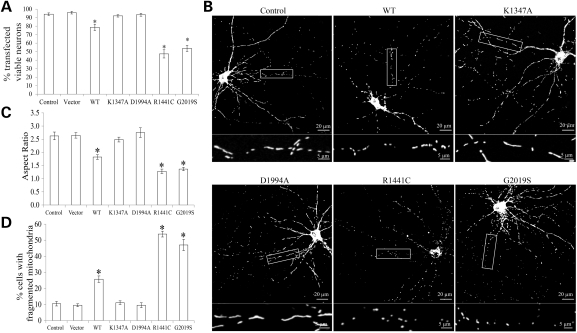
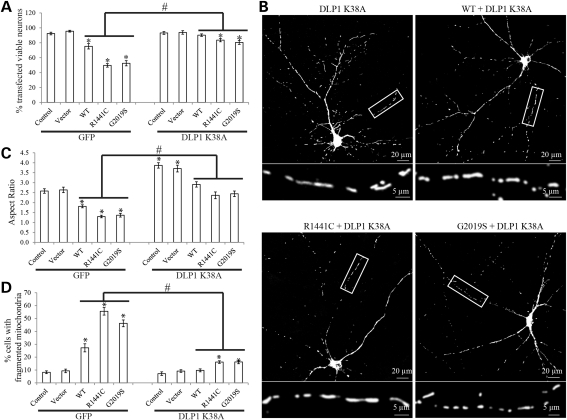
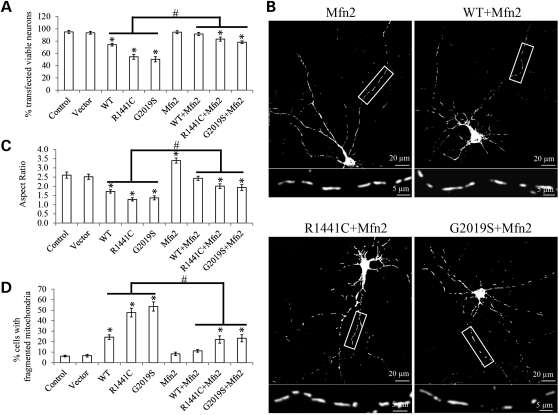
The leucine-rich repeat kinase 2 (LRRK2) mutations are the most common cause of autosomal-dominant Parkinson disease (PD). Mitochondrial dysfunction represents a critical event in the pathogenesis of PD. We demonstrated that wild-type (WT) LRRK2 expression caused mitochondrial fragmentation along with increased mitochondrial dynamin-like protein (DLP1, also known as DRP1), a fission protein, which was further exacerbated by expression of PD-associated mutants (R1441C or G2019S) in both SH-SY5Y and differentiated primary cortical neurons. We also found that LRRK2 interacted with DLP1, and LRRK2-DLP1 interaction was enhanced by PD-associated mutations that probably results in increased mitochondrial DLP1 levels. Co-expression of dominant-negative DLP1 K38A or WT Mfn2 blocked LRRK2-induced mitochondrial fragmentation, mitochondrial dysfunction and neuronal toxicity. Importantly, mitochondrial fragmentation and dysfunction were not observed in cells expressing either GTP-binding deficient mutant LRRK2 K1347A or kinase-dead mutant D1994A which has minimal interaction with DLP1 and did not increase the mitochondrial DLP1 level. We concluded that LRRK2 regulates mitochondrial dynamics by increasing mitochondrial DLP1 through its direct interaction with DLP1, and LRRK2 kinase activity plays a critical role in this process.
Figures
References
-
- Mata I.F., Wedemeyer W.J., Farrer M.J., Taylor J.P., Gallo K.A. LRRK2 in Parkinson's disease: protein domains and functional insights. Trends Neurosci. 2006;29:286–293. - PubMed
-
- Zimprich A., Biskup S., Leitner P., Lichtner P., Farrer M., Lincoln S., Kachergus J., Hulihan M., Uitti R.J., Calne D.B., et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. - PubMed
-
- Paisan-Ruiz C., Jain S., Evans E.W., Gilks W.P., Simon J., van der Brug M., Lopez de Munain A., Aparicio S., Gil A.M., Khan N., et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44:595–600. - PubMed
-
- Lesage S., Brice A. Parkinson's disease: from monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 2009;18:R48–R59. - PubMed
-
- Henchcliffe C., Beal M.F. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat. Clin. Pract. Neurol. 2008;4:600–609. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
