

Activation of endoplasmic reticulum stress by hyperglycemia is essential for Müller cell-derived inflammatory cytokine production in diabetes
- PMID: 22228718
- PMCID: PMC3266398
- DOI: 10.2337/db11-0315
Activation of endoplasmic reticulum stress by hyperglycemia is essential for Müller cell-derived inflammatory cytokine production in diabetes
Abstract
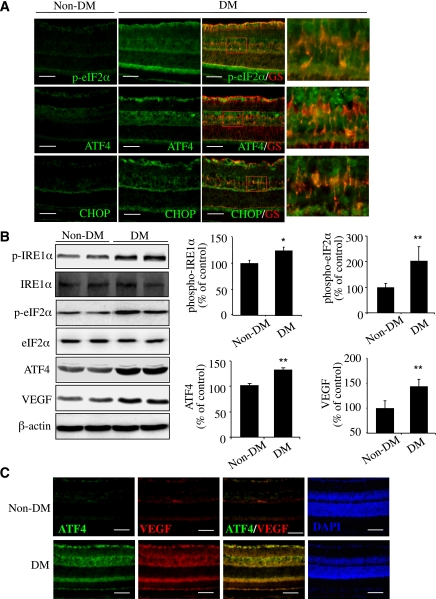
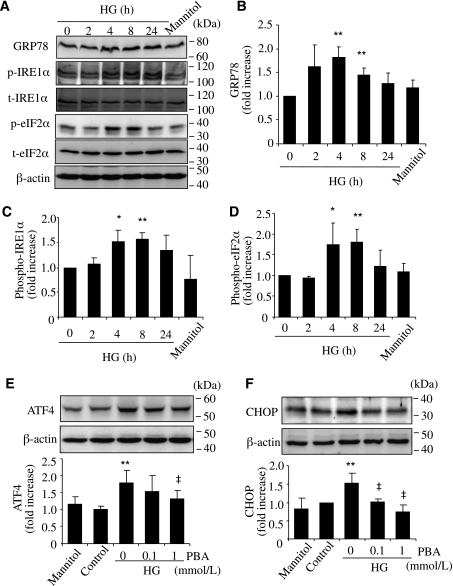
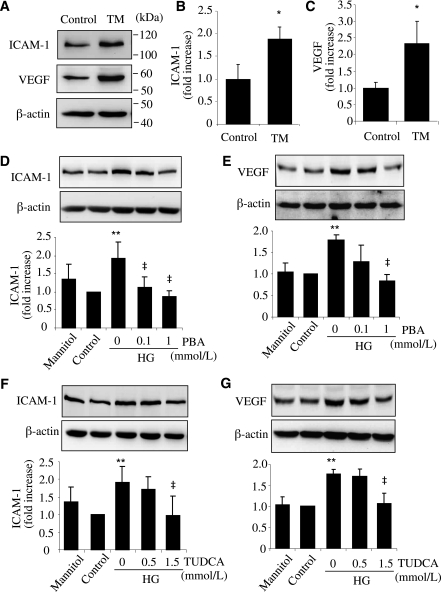
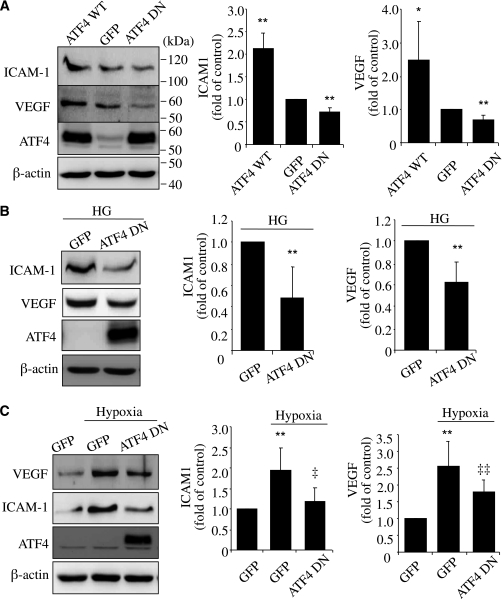
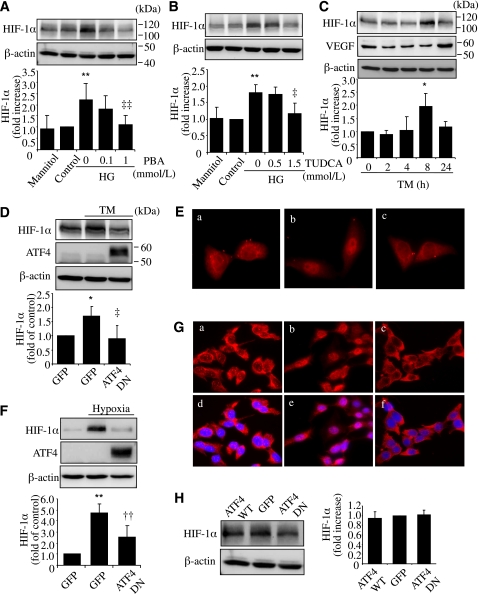
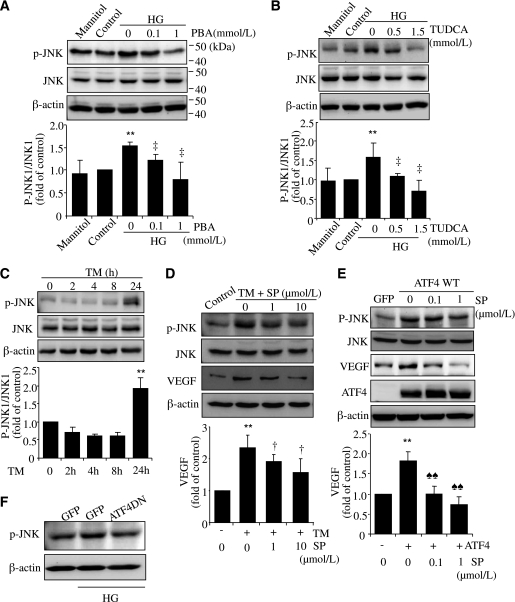
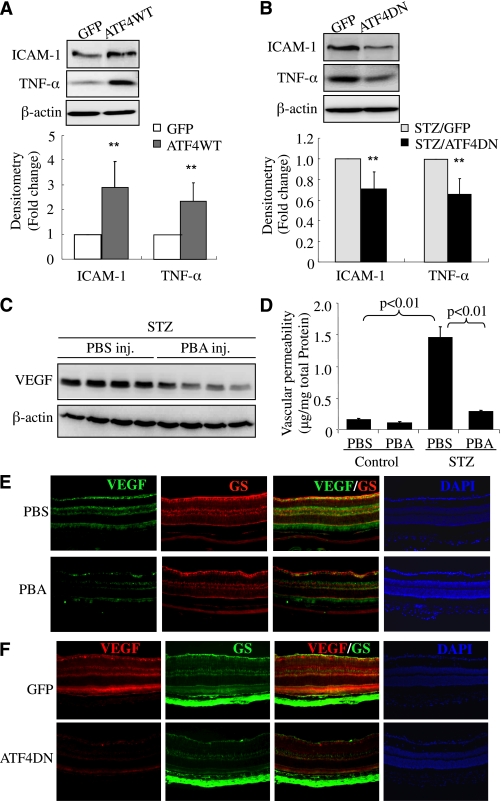
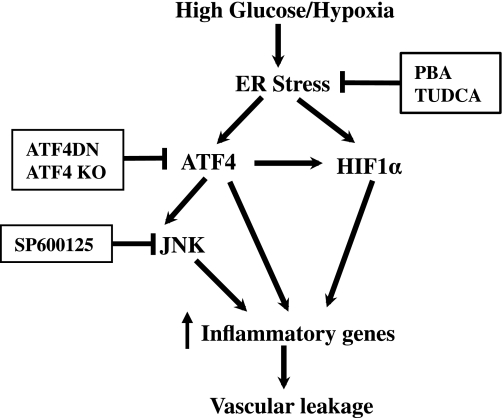
Inflammation plays an important role in diabetes-induced retinal vascular leakage. The purpose of this study is to examine the role of endoplasmic reticulum (ER) stress and the signaling pathway of ER stress-induced activating transcription factor 4 (ATF4) in the regulation of Müller cell-derived inflammatory mediators in diabetic retinopathy. In diabetic animals, elevated ER stress markers, ATF4, and vascular endothelial growth factor (VEGF) expression were partially localized to Müller cells in the retina. In cultured Müller cells, high glucose induced a time-dependent increase of ER stress, ATF4 expression, and inflammatory factor production. Inducing ER stress or overexpressing ATF4 resulted in elevated intracellular adhesion molecule 1 and VEGF proteins in Müller cells. In contrast, alleviation of ER stress or blockade of ATF4 activity attenuated inflammatory gene expression induced by high glucose or hypoxia. Furthermore, we found that ATF4 regulated the c-Jun NH2-terminal kinase pathway resulting in VEGF upregulation. ATF4 was also required for ER stress-induced and hypoxia-inducible factor-1α activation. Finally, we showed that administration of chemical chaperone 4-phenylbutyrate or genetic inhibition of ATF4 successfully attenuated retinal VEGF expression and reduced vascular leakage in mice with STZ-induced diabetes. Taken together, our data indicate that ER stress and ATF4 play a critical role in retinal inflammatory signaling and Müller cell-derived inflammatory cytokine production in diabetes.
Figures
Similar articles
-
Loss of X-box binding protein 1 in Müller cells augments retinal inflammation in a mouse model of diabetes.Diabetologia. 2019 Mar;62(3):531-543. doi: 10.1007/s00125-018-4776-y. Epub 2019 Jan 6. Diabetologia. 2019. PMID: 30612139 Free PMC article.
-
Activating transcription factor 4 mediates hyperglycaemia-induced endothelial inflammation and retinal vascular leakage through activation of STAT3 in a mouse model of type 1 diabetes.Diabetologia. 2012 Sep;55(9):2533-45. doi: 10.1007/s00125-012-2594-1. Epub 2012 Jun 4. Diabetologia. 2012. PMID: 22660795 Free PMC article.
-
Ischaemia-induced retinal neovascularisation and diabetic retinopathy in mice with conditional knockout of hypoxia-inducible factor-1 in retinal Müller cells.Diabetologia. 2011 Jun;54(6):1554-66. doi: 10.1007/s00125-011-2081-0. Epub 2011 Mar 1. Diabetologia. 2011. PMID: 21360191 Free PMC article.
-
Mini review ATF4 and GRP78 as novel molecular targets in ER-Stress modulation for critical COVID-19 patients.Mol Biol Rep. 2022 Feb;49(2):1545-1549. doi: 10.1007/s11033-021-07071-9. Epub 2022 Jan 14. Mol Biol Rep. 2022. PMID: 35028855 Free PMC article. Review.
-
Endoplasmic reticulum stress-related factors protect against diabetic retinopathy.Exp Diabetes Res. 2012;2012:507986. doi: 10.1155/2012/507986. Epub 2011 Dec 10. Exp Diabetes Res. 2012. PMID: 22203836 Free PMC article. Review.
Cited by
-
Heparanase induced by advanced glycation end products (AGEs) promotes macrophage migration involving RAGE and PI3K/AKT pathway.Cardiovasc Diabetol. 2013 Feb 26;12:37. doi: 10.1186/1475-2840-12-37. Cardiovasc Diabetol. 2013. PMID: 23442498 Free PMC article.
-
Molecular mechanisms of diabetic retinopathy: potential therapeutic targets.Middle East Afr J Ophthalmol. 2015 Apr-Jun;22(2):135-44. doi: 10.4103/0974-9233.154386. Middle East Afr J Ophthalmol. 2015. PMID: 25949069 Free PMC article. Review.
-
Regulation of metabolic health by essential dietary amino acids.Mech Ageing Dev. 2019 Jan;177:186-200. doi: 10.1016/j.mad.2018.07.004. Epub 2018 Jul 22. Mech Ageing Dev. 2019. PMID: 30044947 Free PMC article. Review.
-
Palmitic Acid Induces Müller Cell Inflammation that is Potentiated by Co-treatment with Glucose.Sci Rep. 2018 Apr 3;8(1):5459. doi: 10.1038/s41598-018-23601-1. Sci Rep. 2018. PMID: 29626212 Free PMC article.
-
Metabolic Dysregulation and Neurovascular Dysfunction in Diabetic Retinopathy.Antioxidants (Basel). 2020 Dec 8;9(12):1244. doi: 10.3390/antiox9121244. Antioxidants (Basel). 2020. PMID: 33302369 Free PMC article. Review.
References
-
- Joussen AM, Poulaki V, Le ML, et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J 2004;18:1450–1452 - PubMed
-
- Zhang SX, Wang JJ, Gao G, Shao C, Mott R, Ma JX. Pigment epithelium-derived factor (PEDF) is an endogenous antiinflammatory factor. FASEB J 2006;20:323–325 - PubMed
-
- Mizutani M, Gerhardinger C, Lorenzi M. Müller cell changes in human diabetic retinopathy. Diabetes 1998;47:445–449 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
