

Mechanism for release of alkaline phosphatase caused by glycosylphosphatidylinositol deficiency in patients with hyperphosphatasia mental retardation syndrome
- PMID: 22228761
- PMCID: PMC3307314
- DOI: 10.1074/jbc.M111.331090
Mechanism for release of alkaline phosphatase caused by glycosylphosphatidylinositol deficiency in patients with hyperphosphatasia mental retardation syndrome
Abstract
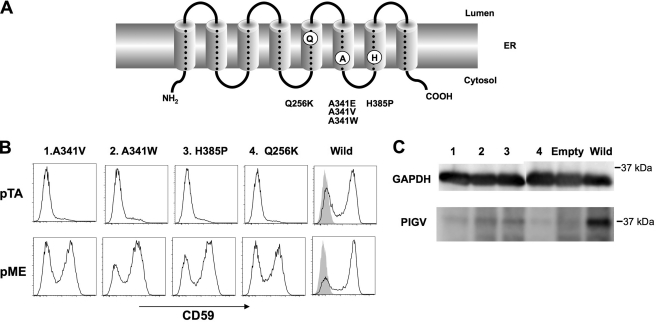
Hyperphosphatasia mental retardation syndrome (HPMR), an autosomal recessive disease characterized by mental retardation and elevated serum alkaline phosphatase (ALP) levels, is caused by mutations in the coding region of the phosphatidylinositol glycan anchor biosynthesis, class V (PIGV) gene, the product of which is a mannosyltransferase essential for glycosylphosphatidylinositol (GPI) biosynthesis. Mutations found in four families caused amino acid substitutions A341E, A341V, Q256K, and H385P, which drastically decreased expression of the PIGV protein. Hyperphosphatasia resulted from secretion of ALP, a GPI-anchored protein normally expressed on the cell surface, into serum due to PIGV deficiency. In contrast, a previously reported PIGM deficiency, in which there is a defect in the transfer of the first mannose, does not result in hyperphosphatasia. To provide insights into the mechanism of ALP secretion in HPMR patients, we took advantage of CHO cell mutants that are defective in various steps of GPI biosynthesis. Secretion of ALP requires GPI transamidase, which in normal cells, cleaves the C-terminal GPI attachment signal peptide and replaces it with GPI. The GPI-anchored protein was secreted substantially into medium from PIGV-, PIGB-, and PIGF-deficient CHO cells, in which incomplete GPI bearing mannose was accumulated. In contrast, ALP was degraded in PIGL-, DPM2-, or PIGX-deficient CHO cells, in which incomplete shorter GPIs that lacked mannose were accumulated. Our results suggest that GPI transamidase recognizes incomplete GPI bearing mannose and cleaves a hydrophobic signal peptide, resulting in secretion of soluble ALP. These results explain the molecular mechanism of hyperphosphatasia in HPMR.
Figures





References
-
- Krawitz P. M., Schweiger M. R., Rödelsperger C., Marcelis C., Kölsch U., Meisel C., Stephani F., Kinoshita T., Murakami Y., Bauer S., Isau M., Fischer A., Dahl A., Kerick M., Hecht J., Köhler S., Jäger M., Grünhagen J., de Condor B. J., Doelken S., Brunner H. G., Meinecke P., Passarge E., Thompson M. D., Cole D. E., Horn D., Roscioli T., Mundlos S., Robinson P. N. (2010) Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat. Genet. 42, 827–829 - PubMed
-
- Kang J. Y., Hong Y., Ashida H., Shishioh N., Murakami Y., Morita Y. S., Maeda Y., Kinoshita T. (2005) PIG-V involved in transferring the second mannose in glycosylphosphatidylinositol. J. Biol. Chem. 280, 9489–9497 - PubMed
-
- Thompson M. D., Nezarati M. M., Gillessen-Kaesbach G., Meinecke P., Mendoza-Londono R., Mornet E., Brun-Heath I., Squarcioni C. P., Legeai-Mallet L., Munnich A., Cole D. E. (2010) Hyperplasia with seizures, neurologic deficit, and characteristic facial features: Five new patients with Mabry syndrome. Am. J. Med. Genet. A 152, 1661–1669 - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
