

Genetics of isolated hypogonadotropic hypogonadism: role of GnRH receptor and other genes
- PMID: 22229029
- PMCID: PMC3249753
- DOI: 10.1155/2012/147893
Genetics of isolated hypogonadotropic hypogonadism: role of GnRH receptor and other genes
Abstract
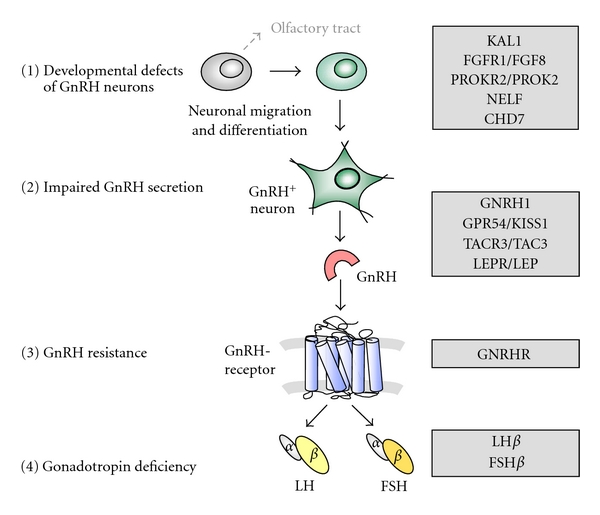
Hypothalamic gonadotropin releasing hormone (GnRH) is a key player in normal puberty and sexual development and function. Genetic causes of isolated hypogonadotropic hypogonadism (IHH) have been identified during the recent years affecting the synthesis, secretion, or action of GnRH. Developmental defects of GnRH neurons and the olfactory bulb are associated with hyposmia, rarely associated with the clinical phenotypes of synkinesia, cleft palate, ear anomalies, or choanal atresia, and may be due to mutations of KAL1, FGFR1/FGF8, PROKR2/PROK2, or CHD7. Impaired GnRH secretion in normosmic patients with IHH may be caused by deficient hypothalamic GPR54/KISS1, TACR3/TAC3, and leptinR/leptin signalling or mutations within the GNRH1 gene itself. Normosmic IHH is predominantly caused by inactivating mutations in the pituitary GnRH receptor inducing GnRH resistance, while mutations of the β-subunits of LH or FSH are very rare. Inheritance of GnRH deficiency may be oligogenic, explaining variable phenotypes. Future research should identify additional genes involved in the complex network of normal and disturbed puberty and reproduction.
Figures

References
-
- De Roux N, Young J, Misrahi M, et al. A family with hypogonadotropic hypogonadism and mutations in the gonadotropin-releasing hormone receptor. New England Journal of Medicine. 1997;337(22):1597–1602. - PubMed
-
- Seminara SB, Messager S, Chatzidaki EE, et al. The GPR54 gene as a regulator of puberty. New England Journal of Medicine. 2003;349(17):1614–1627. - PubMed
-
- Dodé C, Levilliers J, Dupont JM, et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nature Genetics. 2003;33(4):463–465. - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
