

HIV latency
- PMID: 22229121
- PMCID: PMC3234450
- DOI: 10.1101/cshperspect.a007096
HIV latency
Abstract
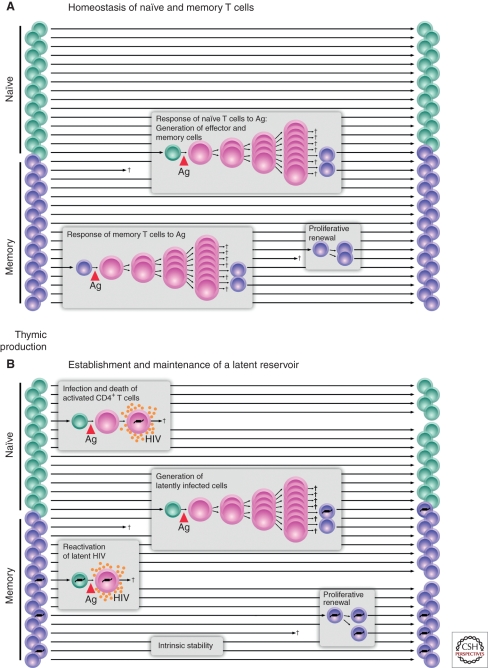
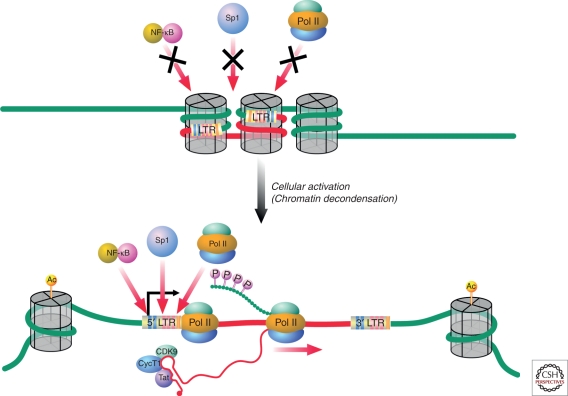
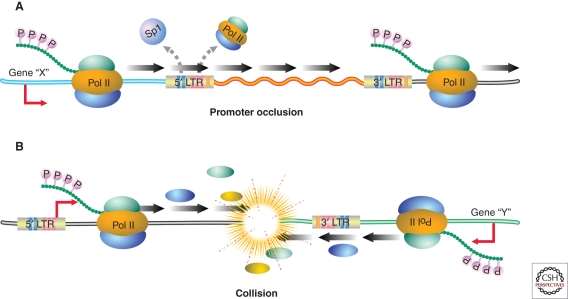
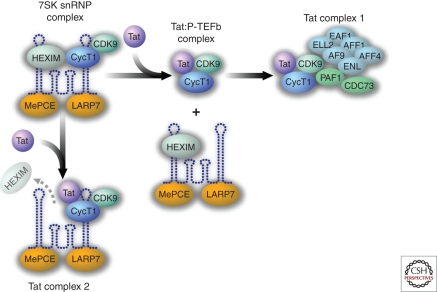
HIV-1 can establish a state of latent infection at the level of individual T cells. Latently infected cells are rare in vivo and appear to arise when activated CD4(+) T cells, the major targets cells for HIV-1, become infected and survive long enough to revert back to a resting memory state, which is nonpermissive for viral gene expression. Because latent virus resides in memory T cells, it persists indefinitely even in patients on potent antiretroviral therapy. This latent reservoir is recognized as a major barrier to curing HIV-1 infection. The molecular mechanisms of latency are complex and include the absence in resting CD4(+) T cells of nuclear forms of key host transcription factors (e.g., NFκB and NFAT), the absence of Tat and associated host factors that promote efficient transcriptional elongation, epigenetic changes inhibiting HIV-1 gene expression, and transcriptional interference. The presence of a latent reservoir for HIV-1 helps explain the presence of very low levels of viremia in patients on antiretroviral therapy. These viruses are released from latently infected cells that have become activated and perhaps from other stable reservoirs but are blocked from additional rounds of replication by the drugs. Several approaches are under exploration for reactivating latent virus with the hope that this will allow elimination of the latent reservoir.
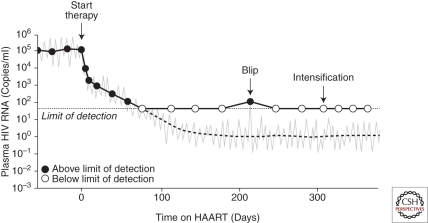
Figures
Similar articles
-
Persistence of wild-type virus and lack of temporal structure in the latent reservoir for human immunodeficiency virus type 1 in pediatric patients with extensive antiretroviral exposure.J Virol. 2002 Sep;76(18):9481-92. doi: 10.1128/jvi.76.18.9481-9492.2002. J Virol. 2002. PMID: 12186930 Free PMC article.
-
Reservoirs for HIV-1: mechanisms for viral persistence in the presence of antiviral immune responses and antiretroviral therapy.Annu Rev Immunol. 2000;18:665-708. doi: 10.1146/annurev.immunol.18.1.665. Annu Rev Immunol. 2000. PMID: 10837072 Review.
-
In vivo analysis of the effect of panobinostat on cell-associated HIV RNA and DNA levels and latent HIV infection.Retrovirology. 2016 May 21;13(1):36. doi: 10.1186/s12977-016-0268-7. Retrovirology. 2016. PMID: 27206407 Free PMC article.
-
From reactivation of latent HIV-1 to elimination of the latent reservoir: the presence of multiple barriers to viral eradication.Bioessays. 2013 Jun;35(6):544-52. doi: 10.1002/bies.201200170. Epub 2013 Apr 24. Bioessays. 2013. PMID: 23613347 Free PMC article. Review.
-
Finding a cure for HIV: will it ever be achievable?J Int AIDS Soc. 2011 Jan 24;14:4. doi: 10.1186/1758-2652-14-4. J Int AIDS Soc. 2011. PMID: 21255462 Free PMC article.
Cited by
-
HIV Excision Utilizing CRISPR/Cas9 Technology: Attacking the Proviral Quasispecies in Reservoirs to Achieve a Cure.MOJ Immunol. 2014 Oct 17;1(4):00022. doi: 10.15406/moji.2014.01.00022. MOJ Immunol. 2014. PMID: 25893217 Free PMC article.
-
Navigating Latency-Inducing Viral Infections: Therapeutic Targeting and Nanoparticle Utilization.Biomater Res. 2024 Oct 16;28:0078. doi: 10.34133/bmr.0078. eCollection 2024. Biomater Res. 2024. PMID: 39416703 Free PMC article. Review.
-
Highly activated p53 contributes to selectively increased apoptosis of latently HIV-1 infected cells upon treatment of anticancer drugs.Virol J. 2016 Aug 16;13(1):141. doi: 10.1186/s12985-016-0595-2. Virol J. 2016. PMID: 27527606 Free PMC article.
-
The Dual-Specificity Kinase DYRK1A Modulates the Levels of Cyclin L2 To Control HIV Replication in Macrophages.J Virol. 2020 Feb 28;94(6):e01583-19. doi: 10.1128/JVI.01583-19. Print 2020 Feb 28. J Virol. 2020. PMID: 31852782 Free PMC article.
-
The Role of the BCL-2 Family of Proteins in HIV-1 Pathogenesis and Persistence.Clin Microbiol Rev. 2019 Oct 30;33(1):e00107-19. doi: 10.1128/CMR.00107-19. Print 2019 Dec 18. Clin Microbiol Rev. 2019. PMID: 31666279 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials