

Structure of a KirBac potassium channel with an open bundle crossing indicates a mechanism of channel gating
- PMID: 22231399
- PMCID: PMC3272479
- DOI: 10.1038/nsmb.2208
Structure of a KirBac potassium channel with an open bundle crossing indicates a mechanism of channel gating
Abstract
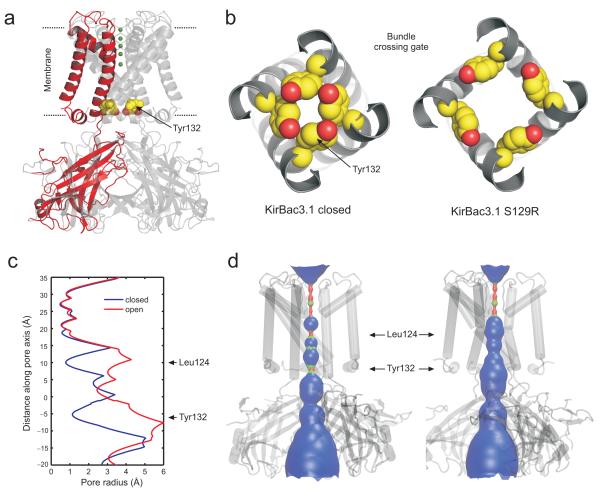
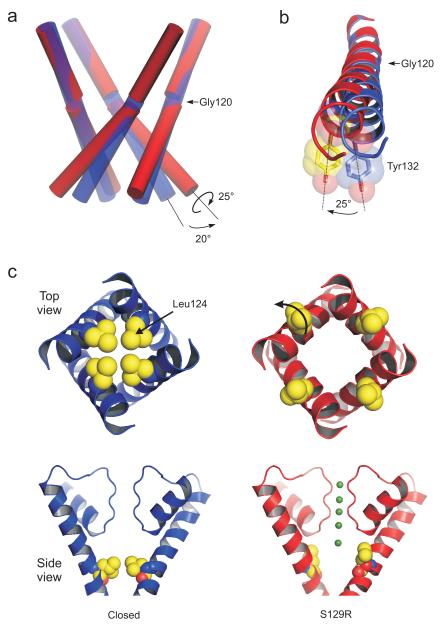
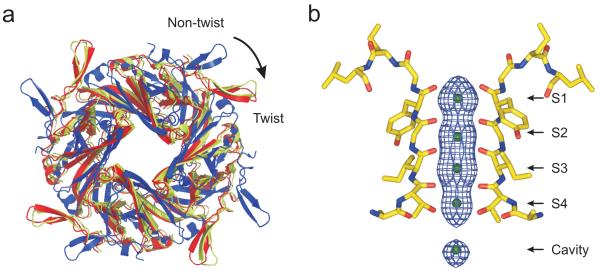
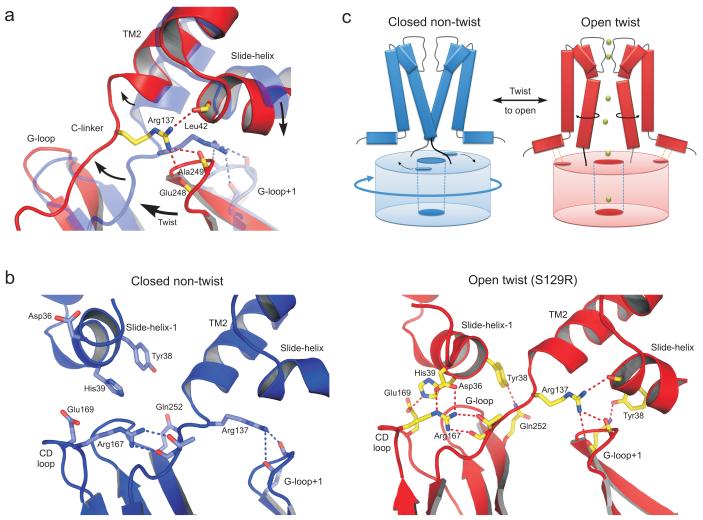
KirBac channels are prokaryotic homologs of mammalian inwardly rectifying (Kir) potassium channels, and recent crystal structures of both Kir and KirBac channels have provided major insight into their unique structural architecture. However, all of the available structures are closed at the helix bundle crossing, and therefore the structural mechanisms that control opening of their primary activation gate remain unknown. In this study, we engineered the inner pore-lining helix (TM2) of KirBac3.1 to trap the bundle crossing in an apparently open conformation and determined the crystal structure of this mutant channel to 3.05 Å resolution. Contrary to previous speculation, this new structure suggests a mechanistic model in which rotational 'twist' of the cytoplasmic domain is coupled to opening of the bundle-crossing gate through a network of inter- and intrasubunit interactions that involve the TM2 C-linker, slide helix, G-loop and the CD loop.
Figures
References
-
- Hibino H, et al. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev. 2010;90:291–366. - PubMed
-
- Bichet D, Haass FA, Jan LY. Merging functional studies with structures of inward-rectifier K+ channels. Nat Rev Neurosci. 2003;4:957–67. - PubMed
-
- Swartz KJ. Towards a structural view of gating in potassium channels. Nat Rev Neurosci. 2004;5:905–16. - PubMed
-
- Kuo A, et al. Crystal structure of the potassium channel KirBac1.1 in the closed state. Science. 2003;300:1922–6. - PubMed
-
- Kuo A, Domene C, Johnson LN, Doyle DA, Venien-Bryan C. Two different conformational states of the KirBac3.1 potassium channel revealed by electron crystallography. Structure. 2005;13:1463–72. - PubMed
Additional references for methods section
-
- Collaborative Computational Project, Number 4 The CCP4 Suite: Programs for Protein Crystallography. Acta Cryst D. 1994;50:760–763. - PubMed
-
- Evans PR. Scaling and assessment of data quality. Acta Cryst D. 2006;62:82–82. - PubMed
-
- Blanc E, et al. Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Cryst D. 2004;60:2210–2221. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
- BB/H007296/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BBS/B/16011/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/F013035/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BEP17032/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- B19456/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
