

Systems-based approaches to cardiovascular disease
- PMID: 22231714
- PMCID: PMC3823242
- DOI: 10.1038/nrcardio.2011.208
Systems-based approaches to cardiovascular disease
Abstract
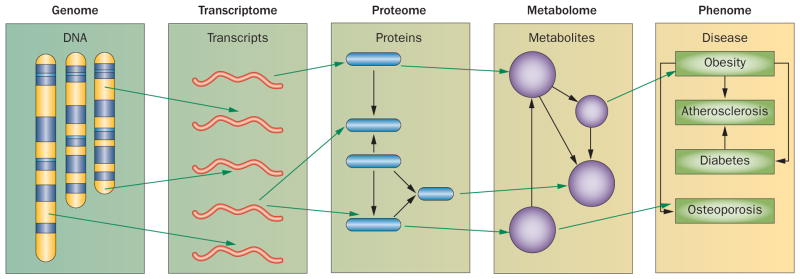
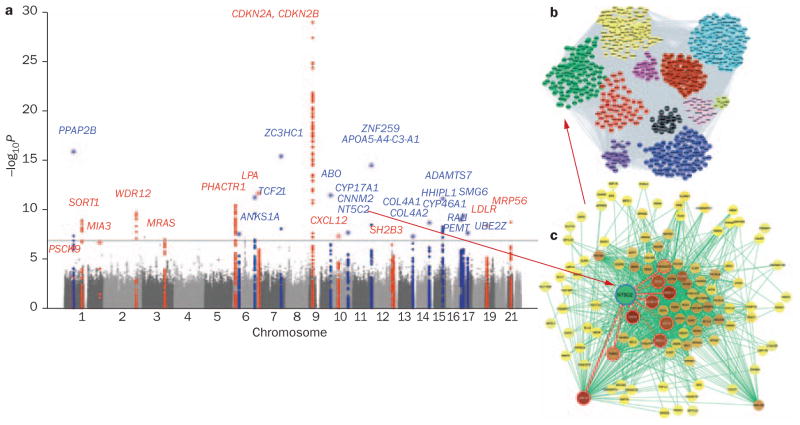
Common cardiovascular diseases, such as atherosclerosis and congestive heart failure, are exceptionally complex, involving a multitude of environmental and genetic factors that often show nonlinear interactions as well as being highly dependent on sex, age, and even the maternal environment. Although focused, reductionistic approaches have led to progress in elucidating the pathophysiology of cardiovascular diseases, such approaches are poorly powered to address complex interactions. Over the past decade, technological advances have made it possible to interrogate biological systems on a global level, raising hopes that, in combination with computational approaches, it may be possible to more fully address the complexities of cardiovascular diseases. In this Review, we provide an overview of such systems-based approaches to cardiovascular disease and discuss their translational implications.
Conflict of interest statement
The authors declare no competing interests.
Figures
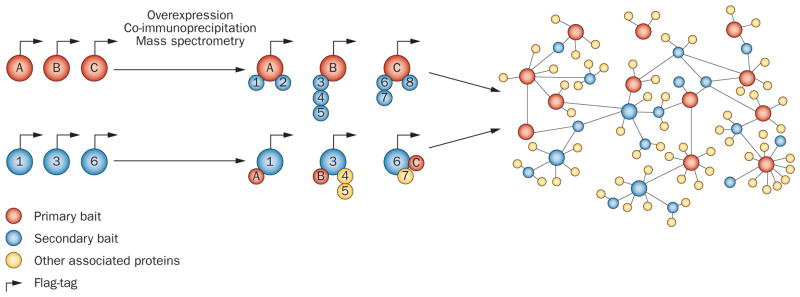
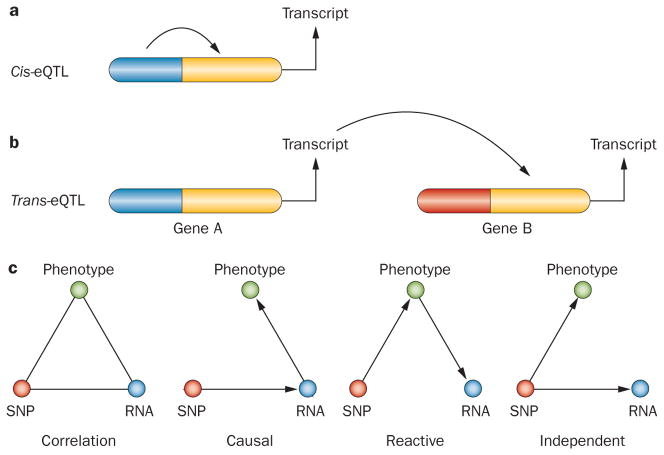
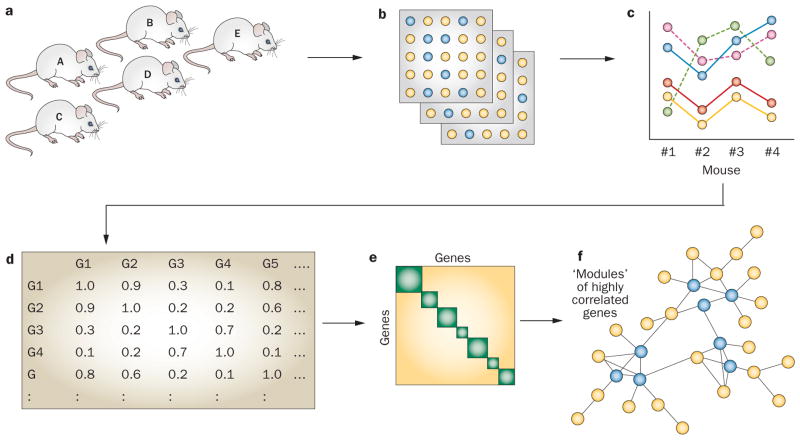
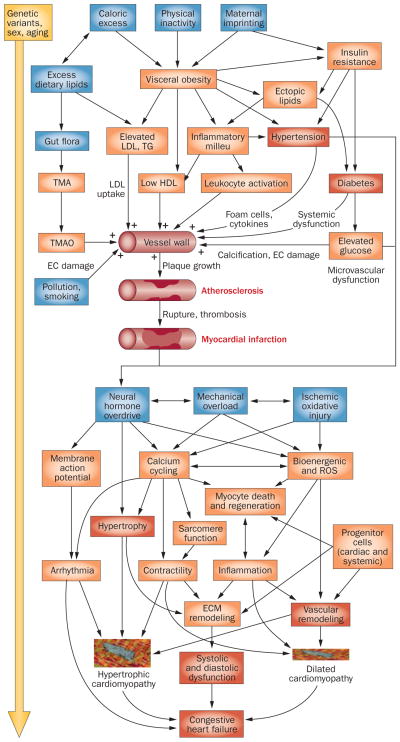
References
-
- Barabasi AL. Linked: The new science of networks. Penguin Books; London: 2002.
-
- Ideker T, Galitski T, Hood L. A new approach to decoding life: systems biology. Annu Rev Genomics Hum Genet. 2001;2:343–372. - PubMed
-
- Nadeau JH, Subramaniam S. Systems biology and medicine: a new take on an old paradigm. Wiley Interdiscip Rev Syst Biol Med. 2009;1:1–3. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
