

Mass spectrometry-based proteomics: qualitative identification to activity-based protein profiling
- PMID: 22231900
- PMCID: PMC3288153
- DOI: 10.1002/wsbm.166
Mass spectrometry-based proteomics: qualitative identification to activity-based protein profiling
Abstract
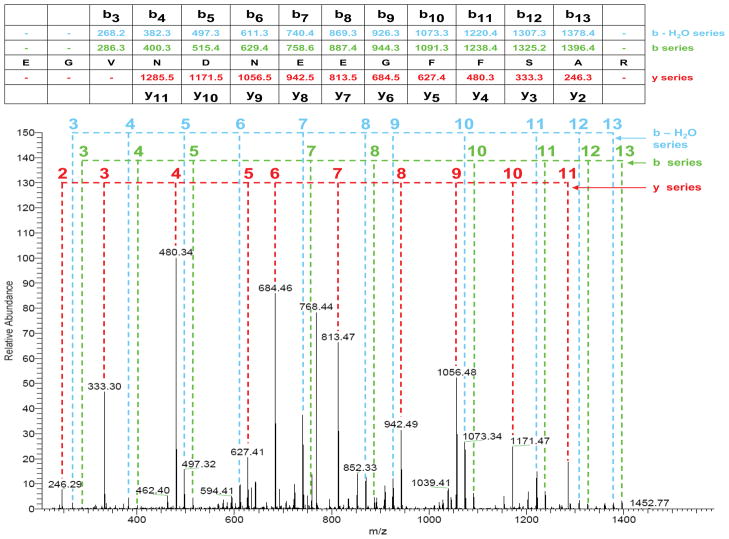
Mass spectrometry has become the method of choice for proteome characterization, including multicomponent protein complexes (typically tens to hundreds of proteins) and total protein expression (up to tens of thousands of proteins), in biological samples. Qualitative sequence assignment based on MS/MS spectra is relatively well-defined, while statistical metrics for relative quantification have not completely stabilized. Nonetheless, proteomics studies have progressed to the point whereby various gene-, pathway-, or network-oriented computational frameworks may be used to place mass spectrometry data into biological context. Despite this progress, the dynamic range of protein expression remains a significant hurdle, and impedes comprehensive proteome analysis. Methods designed to enrich specific protein classes have emerged as an effective means to characterize enzymes or other catalytically active proteins that are otherwise difficult to detect in typical discovery mode proteomics experiments. Collectively, these approaches will facilitate identification of biomarkers and pathways relevant to diagnosis and treatment of human disease.
Copyright © 2012 Wiley Periodicals, Inc.
Figures






References
-
- Weston AD, Hood L. Systems biology, proteomics, and the future of health care: toward predictive, preventative, and personalized medicine. J Proteome Res. 2004;3(2):179–96. - PubMed
-
- Hu Q, et al. The Orbitrap: a new mass spectrometer. J Mass Spectrom. 2005;40(4):430–443. - PubMed
-
- Loboda AV, et al. A tandem quadrupole/time-of-flight mass spectrometer with a matrix-assisted laser desorption/ionization source: design and performance. Rapid Commun Mass Spectrom. 2000;14(12):1047–57. - PubMed
-
- Wenzel RJ, et al. Analysis of megadalton ions using cryodetection MALDI time-of-flight mass spectrometry. Anal Chem. 2005;77(14):4329–37. - PubMed
-
- Zhou M, Robinson CV. When proteomics meets structural biology. Trends Biochem Sci. 2010;35(9):522–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
