

Complete OATP1B1 and OATP1B3 deficiency causes human Rotor syndrome by interrupting conjugated bilirubin reuptake into the liver
- PMID: 22232210
- PMCID: PMC3266790
- DOI: 10.1172/JCI59526
Complete OATP1B1 and OATP1B3 deficiency causes human Rotor syndrome by interrupting conjugated bilirubin reuptake into the liver
Abstract
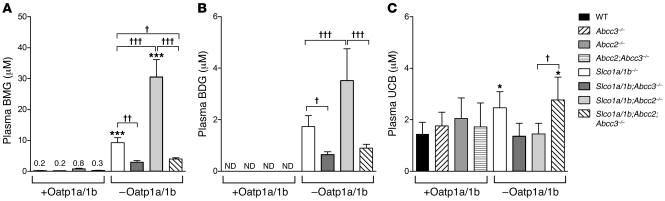
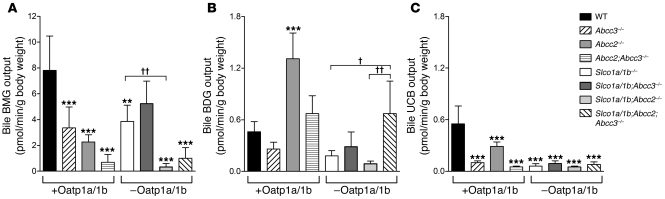
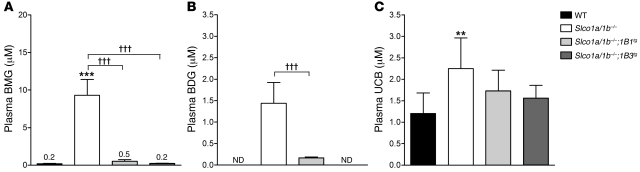
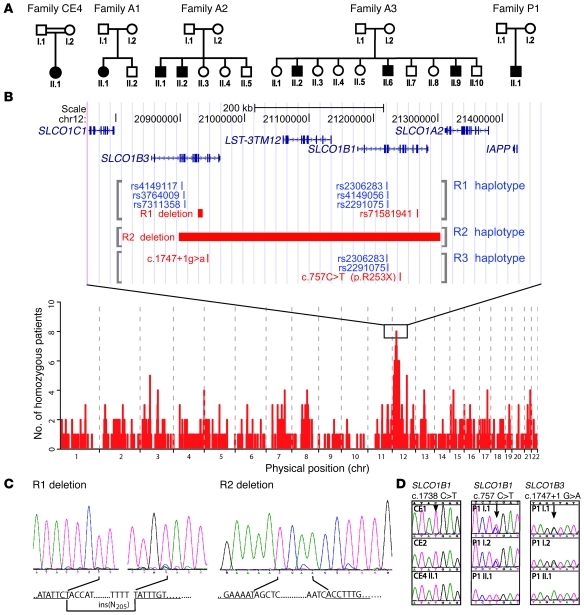
Bilirubin, a breakdown product of heme, is normally glucuronidated and excreted by the liver into bile. Failure of this system can lead to a buildup of conjugated bilirubin in the blood, resulting in jaundice. The mechanistic basis of bilirubin excretion and hyperbilirubinemia syndromes is largely understood, but that of Rotor syndrome, an autosomal recessive disorder characterized by conjugated hyperbilirubinemia, coproporphyrinuria, and near-absent hepatic uptake of anionic diagnostics, has remained enigmatic. Here, we analyzed 8 Rotor-syndrome families and found that Rotor syndrome was linked to mutations predicted to cause complete and simultaneous deficiencies of the organic anion transporting polypeptides OATP1B1 and OATP1B3. These important detoxification-limiting proteins mediate uptake and clearance of countless drugs and drug conjugates across the sinusoidal hepatocyte membrane. OATP1B1 polymorphisms have previously been linked to drug hypersensitivities. Using mice deficient in Oatp1a/1b and in the multispecific sinusoidal export pump Abcc3, we found that Abcc3 secretes bilirubin conjugates into the blood, while Oatp1a/1b transporters mediate their hepatic reuptake. Transgenic expression of human OATP1B1 or OATP1B3 restored the function of this detoxification-enhancing liver-blood shuttle in Oatp1a/1b-deficient mice. Within liver lobules, this shuttle may allow flexible transfer of bilirubin conjugates (and probably also drug conjugates) formed in upstream hepatocytes to downstream hepatocytes, thereby preventing local saturation of further detoxification processes and hepatocyte toxic injury. Thus, disruption of hepatic reuptake of bilirubin glucuronide due to coexisting OATP1B1 and OATP1B3 deficiencies explains Rotor-type hyperbilirubinemia. Moreover, OATP1B1 and OATP1B3 null mutations may confer substantial drug toxicity risks.
Figures
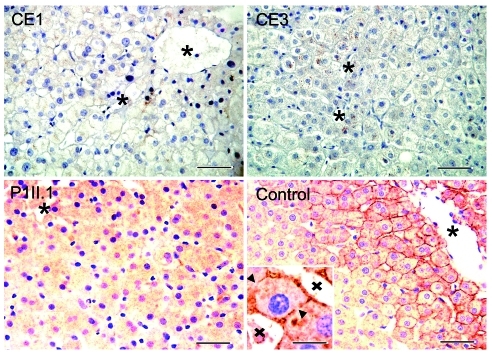
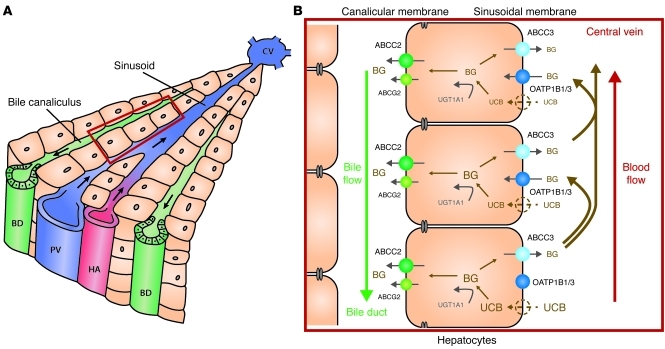
Comment in
-
Hereditary conjugated hyperbilirubinaemia: 37 years later.J Hepatol. 2013 Feb;58(2):388-90. doi: 10.1016/j.jhep.2012.08.025. Epub 2012 Sep 11. J Hepatol. 2013. PMID: 22982575
Similar articles
-
Hereditary conjugated hyperbilirubinaemia: 37 years later.J Hepatol. 2013 Feb;58(2):388-90. doi: 10.1016/j.jhep.2012.08.025. Epub 2012 Sep 11. J Hepatol. 2013. PMID: 22982575
-
New insights in bilirubin metabolism and their clinical implications.World J Gastroenterol. 2013 Oct 14;19(38):6398-407. doi: 10.3748/wjg.v19.i38.6398. World J Gastroenterol. 2013. PMID: 24151358 Free PMC article. Review.
-
The roles of MRP2, MRP3, OATP1B1, and OATP1B3 in conjugated hyperbilirubinemia.Drug Metab Dispos. 2014 Apr;42(4):561-5. doi: 10.1124/dmd.113.055772. Epub 2014 Jan 23. Drug Metab Dispos. 2014. PMID: 24459177 Review.
-
OATP1B1/1B3 deficiency exacerbates hyperbilirubinemia in erythropoietic protoporphyria.Drug Metab Dispos. 2025 Jul;53(7):100105. doi: 10.1016/j.dmd.2025.100105. Epub 2025 May 27. Drug Metab Dispos. 2025. PMID: 40540978
-
Influence of human OATP1B1, OATP1B3, and OATP1A2 on the pharmacokinetics of methotrexate and paclitaxel in humanized transgenic mice.Clin Cancer Res. 2013 Feb 15;19(4):821-32. doi: 10.1158/1078-0432.CCR-12-2080. Epub 2012 Dec 14. Clin Cancer Res. 2013. PMID: 23243220
Cited by
-
Coproporphyrin I Can Serve as an Endogenous Biomarker for OATP1B1 Inhibition: Assessment Using a Glecaprevir/Pibrentasvir Clinical Study.Clin Transl Sci. 2021 Jan;14(1):373-381. doi: 10.1111/cts.12888. Epub 2020 Oct 24. Clin Transl Sci. 2021. PMID: 33048456 Free PMC article. Clinical Trial.
-
A study on setting standards for near-infrared fluorescence-image guided surgery (NIRFGS) time lapse monitoring based on preoperative liver function assessment.Ann Transl Med. 2022 Jan;10(2):96. doi: 10.21037/atm-21-6975. Ann Transl Med. 2022. PMID: 35282106 Free PMC article.
-
Protein-protein interactions of drug uptake transporters that are important for liver and kidney.Biochem Pharmacol. 2019 Oct;168:384-391. doi: 10.1016/j.bcp.2019.07.026. Epub 2019 Aug 2. Biochem Pharmacol. 2019. PMID: 31381872 Free PMC article. Review.
-
Quantitative Kinetic Models from Intravital Microscopy: A Case Study Using Hepatic Transport.J Phys Chem B. 2019 Aug 29;123(34):7302-7312. doi: 10.1021/acs.jpcb.9b04729. Epub 2019 Aug 15. J Phys Chem B. 2019. PMID: 31298856 Free PMC article.
-
Association of neonatal hyperbilirubinemia in breast-fed infants with UGT1A1 or SLCOs polymorphisms.J Hum Genet. 2015 Jan;60(1):35-40. doi: 10.1038/jhg.2014.98. Epub 2014 Nov 13. J Hum Genet. 2015. PMID: 25391605
References
-
- Chowdhury JR, Chowdhury NR, Jansen PLM. Bilirubin metabolism and its disorders. In: Boyer TD, Wright TL, Manns MP, Zakim D, eds.Zakim and Boyer’s Hepatology. A Textbook of Liver Diseases . Vol. 2. Philadelphia, Pennsylvania, USA: Saunders Elsevier; 2006:1449–1474.
-
- Chowdhury JR, Wolkoff AW, Chowdhury NR, Arias IM. Hereditary jaundice and disorders of bilirubin metabolism. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds.The Metabolic and Molecular Bases of Inherited Disease . Vol. 2. New York, New York, USA: McGraw Hill; 2001:3063–3101.
-
- Rotor AB, Manahan L, Florentin A. Familial nonhemolytic jaundice with direct van den Bergh reaction. Acta Med Phil. 1948;5:37–49.
-
- Bar-Meir S, Baron J, Seligson U, Gottesfeld F, Levy R, Gilat T. 99mTc-HIDA cholescintigraphy in Dubin-Johnson and Rotor syndromes. Radiology. 1982;142(3):743–746. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
