

Domain architecture of a calcium-permeable AMPA receptor in a ligand-free conformation
- PMID: 22232575
- PMCID: PMC3249379
- DOI: 10.3389/fnmol.2011.00056
Domain architecture of a calcium-permeable AMPA receptor in a ligand-free conformation
Abstract
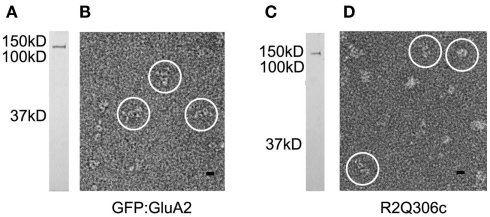
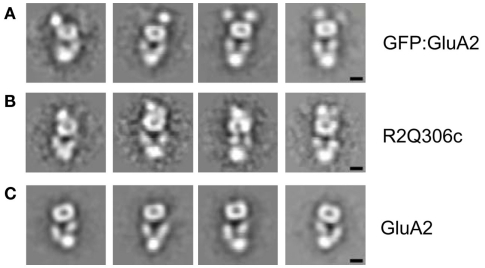
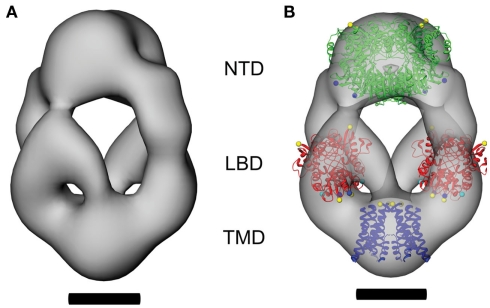
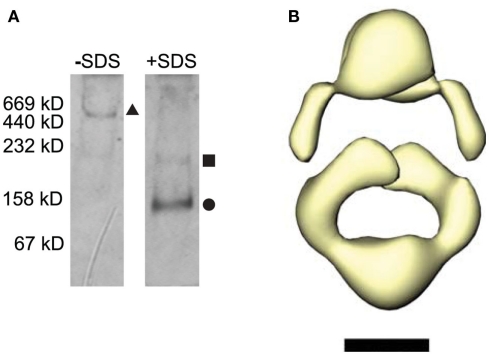
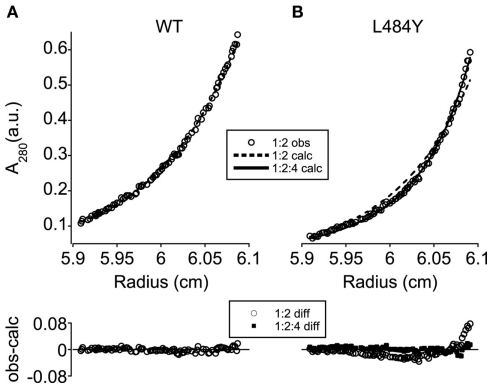
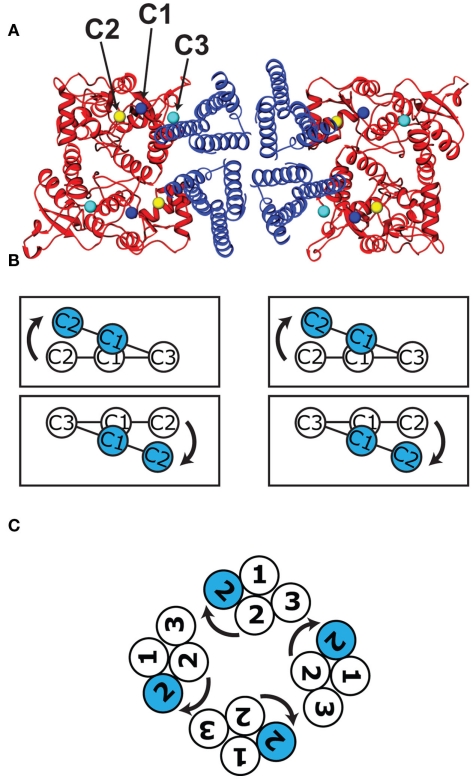
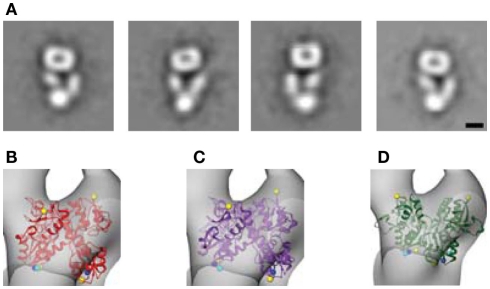
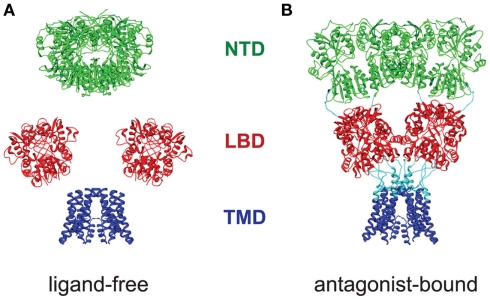
Ligand-gated ion channels couple the free energy of agonist binding to the gating of selective transmembrane ion pores, permitting cells to regulate ion flux in response to external chemical stimuli. However, the stereochemical mechanisms responsible for this coupling remain obscure. In the case of the ionotropic glutamate receptors (iGluRs), the modular nature of receptor subunits has facilitated structural analysis of the N-terminal domain (NTD), and of multiple conformations of the ligand-binding domain (LBD). Recently, the crystallographic structure of an antagonist-bound form of the receptor was determined. However, disulfide trapping of this conformation blocks channel opening, suggesting that channel activation involves additional quaternary packing arrangements. To explore the conformational space available to iGluR channels, we report here a second, clearly distinct domain architecture of homotetrameric, calcium-permeable AMPA receptors, determined by single-particle electron microscopy of untagged and fluorescently tagged constructs in a ligand-free state. It reveals a novel packing of NTD dimers, and a separation of LBD dimers across a central vestibule. In this arrangement, which reconciles diverse functional observations, agonist-induced cleft closure across LBD dimers can be converted into a twisting motion that provides a basis for receptor activation.
Keywords: electron microscopy; ionotropic glutamate receptors; ligand-gated ion channels; protein structure; receptor activation; single-particle reconstruction.
Figures
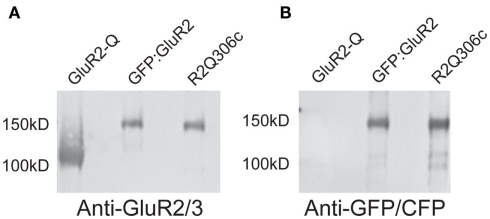
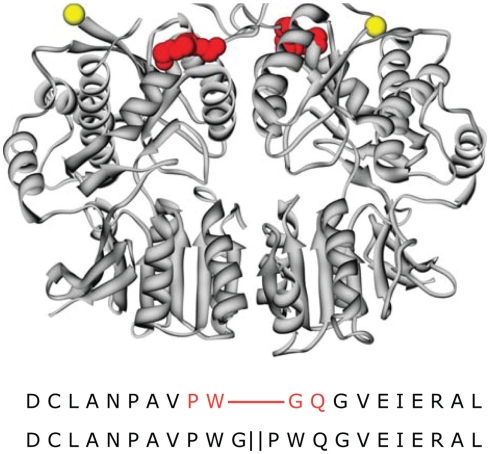
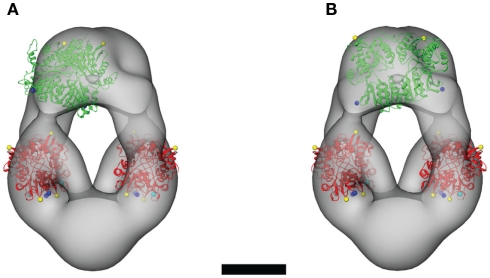
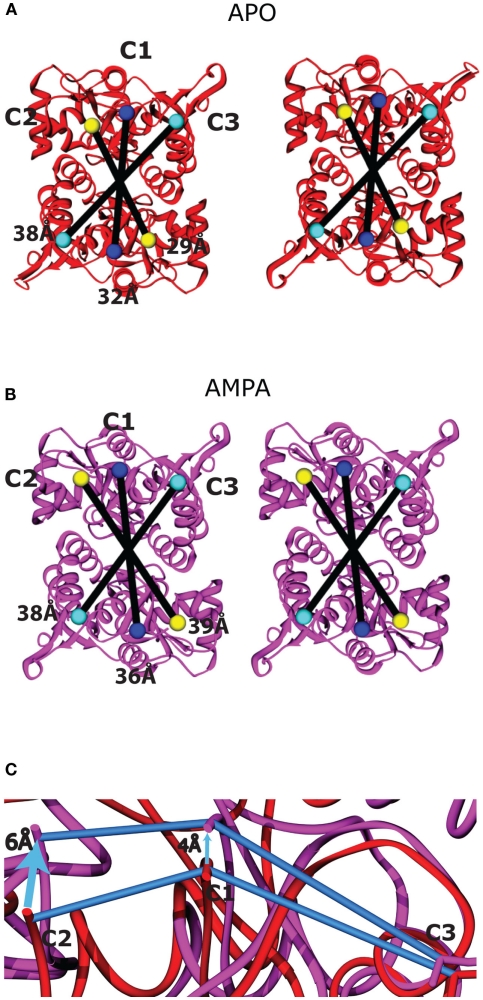
Similar articles
-
Glutamate receptor desensitization is mediated by changes in quaternary structure of the ligand binding domain.Proc Natl Acad Sci U S A. 2013 Apr 9;110(15):5921-6. doi: 10.1073/pnas.1217549110. Epub 2013 Mar 25. Proc Natl Acad Sci U S A. 2013. PMID: 23530186 Free PMC article.
-
Gating Motions and Stationary Gating Properties of Ionotropic Glutamate Receptors: Computation Meets Electrophysiology.Acc Chem Res. 2017 Apr 18;50(4):814-822. doi: 10.1021/acs.accounts.6b00598. Epub 2017 Feb 10. Acc Chem Res. 2017. PMID: 28186717 Free PMC article. Review.
-
Enhanced efficacy without further cleft closure: reevaluating twist as a source of agonist efficacy in AMPA receptors.J Neurosci. 2010 Jan 27;30(4):1463-70. doi: 10.1523/JNEUROSCI.4558-09.2010. J Neurosci. 2010. PMID: 20107073 Free PMC article.
-
Structure, Dynamics, and Allosteric Potential of Ionotropic Glutamate Receptor N-Terminal Domains.Biophys J. 2015 Sep 15;109(6):1136-48. doi: 10.1016/j.bpj.2015.06.061. Epub 2015 Aug 6. Biophys J. 2015. PMID: 26255587 Free PMC article. Review.
-
Emerging issues of connexin channels: biophysics fills the gap.Q Rev Biophys. 2001 Aug;34(3):325-472. doi: 10.1017/s0033583501003705. Q Rev Biophys. 2001. PMID: 11838236 Review.
Cited by
-
Expansion microscopy: A powerful nanoscale imaging tool for neuroscientists.Neurobiol Dis. 2021 Jul;154:105362. doi: 10.1016/j.nbd.2021.105362. Epub 2021 Apr 2. Neurobiol Dis. 2021. PMID: 33813047 Free PMC article. Review.
-
Structure and gating of tetrameric glutamate receptors.J Physiol. 2015 Jan 1;593(1):29-38. doi: 10.1113/jphysiol.2013.264911. Epub 2013 Nov 25. J Physiol. 2015. PMID: 25556785 Free PMC article. Review.
-
The neurotransmitter glutamate and human T cells: glutamate receptors and glutamate-induced direct and potent effects on normal human T cells, cancerous human leukemia and lymphoma T cells, and autoimmune human T cells.J Neural Transm (Vienna). 2014 Aug;121(8):983-1006. doi: 10.1007/s00702-014-1167-5. Epub 2014 Mar 2. J Neural Transm (Vienna). 2014. PMID: 24584970 Review.
-
Retour aux sources: defining the structural basis of glutamate receptor activation.J Physiol. 2015 Jan 1;593(1):97-110. doi: 10.1113/jphysiol.2014.277921. Epub 2014 Oct 21. J Physiol. 2015. PMID: 25556791 Free PMC article. Review.
-
Glutamate receptor desensitization is mediated by changes in quaternary structure of the ligand binding domain.Proc Natl Acad Sci U S A. 2013 Apr 9;110(15):5921-6. doi: 10.1073/pnas.1217549110. Epub 2013 Mar 25. Proc Natl Acad Sci U S A. 2013. PMID: 23530186 Free PMC article.
References
LinkOut - more resources
Full Text Sources