

Axon degeneration: molecular mechanisms of a self-destruction pathway
- PMID: 22232700
- PMCID: PMC3255986
- DOI: 10.1083/jcb.201108111
Axon degeneration: molecular mechanisms of a self-destruction pathway
Abstract
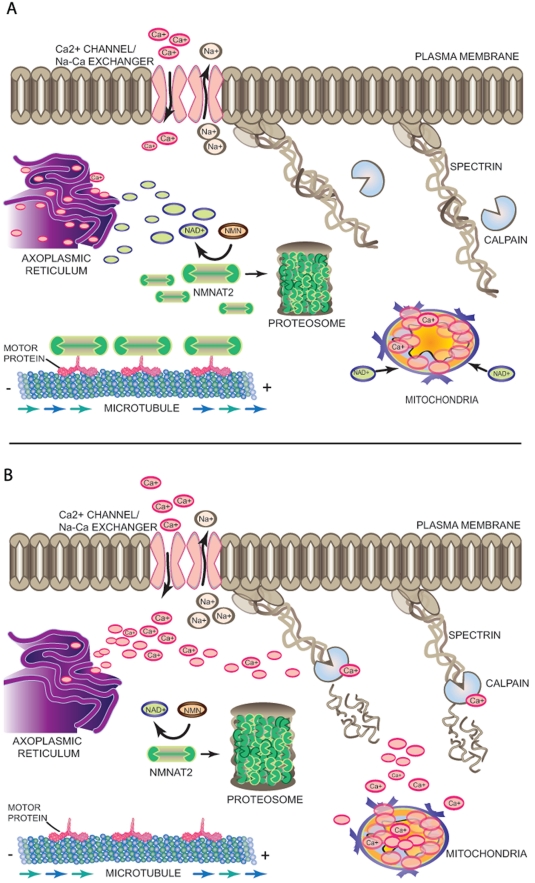
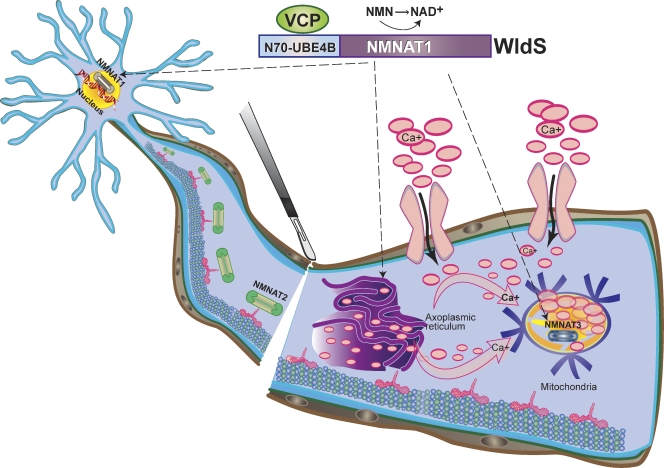
Axon degeneration is a characteristic event in many neurodegenerative conditions including stroke, glaucoma, and motor neuropathies. However, the molecular pathways that regulate this process remain unclear. Axon loss in chronic neurodegenerative diseases share many morphological features with those in acute injuries, and expression of the Wallerian degeneration slow (WldS) transgene delays nerve degeneration in both events, indicating a common mechanism of axonal self-destruction in traumatic injuries and degenerative diseases. A proposed model of axon degeneration is that nerve insults lead to impaired delivery or expression of a local axonal survival factor, which results in increased intra-axonal calcium levels and calcium-dependent cytoskeletal breakdown.
Figures

References
-
- Adalbert R., Gillingwater T.H., Haley J.E., Bridge K., Beirowski B., Berek L., Wagner D., Grumme D., Thomson D., Celik A., et al. 2005. A rat model of slow Wallerian degeneration (WldS) with improved preservation of neuromuscular synapses. Eur. J. Neurosci. 21:271–277 10.1111/j.1460-9568.2004.03833.x - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
