

Disruptions in the regulation of extracellular glutamate by neurons and glia in the rat striatum two days after diffuse brain injury
- PMID: 22233432
- PMCID: PMC3325551
- DOI: 10.1089/neu.2011.2261
Disruptions in the regulation of extracellular glutamate by neurons and glia in the rat striatum two days after diffuse brain injury
Abstract
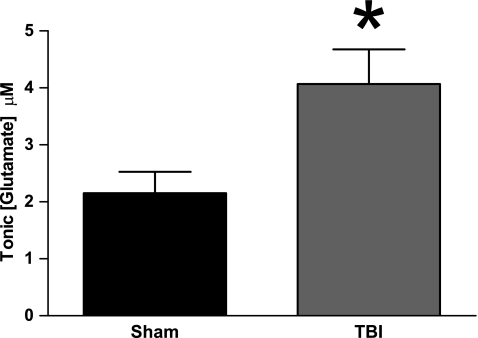
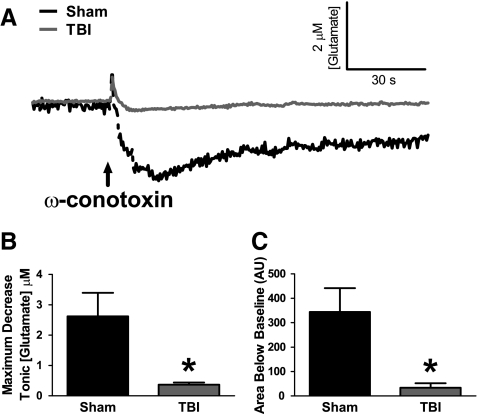
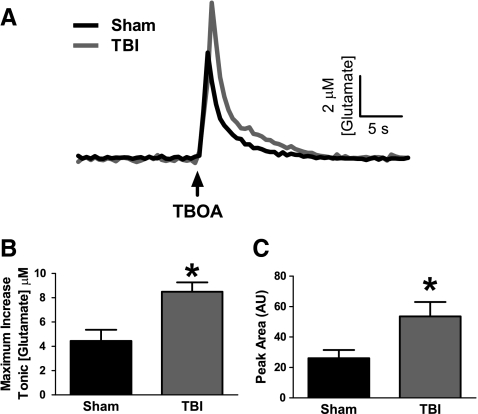
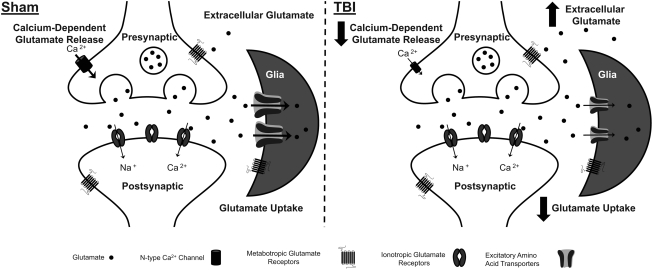
Disrupted regulation of extracellular glutamate in the central nervous system contributes to and can exacerbate the acute pathophysiology of traumatic brain injury (TBI). Previously, we reported increased extracellular glutamate in the striatum of anesthetized rats 2 days after diffuse brain injury. To determine the mechanism(s) responsible for increased extracellular glutamate, we used enzyme-based microelectrode arrays (MEAs) coupled with specific pharmacological agents targeted at in vivo neuronal and glial regulation of extracellular glutamate. After TBI, extracellular glutamate was significantly increased in the striatum by (∼90%) averaging 4.1±0.6 μM compared with sham 2.2±0.4 μM. Calcium-dependent neuronal glutamate release, investigated by local application of an N-type calcium channel blocker, was no longer a significant source of extracellular glutamate after TBI, compared with sham. In brain-injured animals, inhibition of glutamate uptake with local application of an excitatory amino acid transporter inhibitor produced significantly greater increase in glutamate spillover (∼ 65%) from the synapses compared with sham. Furthermore, glutamate clearance measured by locally applying glutamate into the extracellular space revealed significant reductions in glutamate clearance parameters in brain-injured animals compared with sham. Taken together, these data indicate that disruptions in calcium-mediated glutamate release and glial regulation of extracellular glutamate contribute to increased extracellular glutamate in the striatum 2 days after diffuse brain injury. Overall, these data suggest that therapeutic strategies used to regulate glutamate release and uptake may improve excitatory circuit function and, possibly, outcomes following TBI.
Figures

References
-
- Albrecht P. Lewerenz J. Dittmer S. Noack R. Maher P. Methner A. Mechanisms of oxidative glutamate toxicity: the glutamate/cystine antiporter system xc- as a neuroprotective drug target. CNS Neurol. Disord. Drug Targets. 2010;9:373–382. - PubMed
-
- Allen J.W. Ivanova S.A. Fan L. Espey M.G. Basile A.S. Faden A.I. Group II metabotropic glutamate receptor activation attenuates traumatic neuronal injury and improves neurological recovery after traumatic brain injury. J. Pharmacol. Exp. Ther. 1999;290:112–120. - PubMed
-
- Battaglia G. Monn J.A. Schoepp D.D. In vivo inhibition of veratridine-evoked release of striatal excitatory amino acids by the group II metabotropic glutamate receptor agonist LY354740 in rats. Neurosci. Lett. 1997;229:161–164. - PubMed
-
- Bond A. Jones N.M. Hicks C.A. Whiffin G.M. Ward M.A. O'Neill M.F. Kingston A.E. Monn J.A. Ornstein P.L. Schoepp D.D. Lodge D. O'Neill M.J. Neuroprotective effects of LY379268, a selective mGlu2/3 receptor agonist: investigations into possible mechanism of action in vivo. J. Pharmacol. Exp. Ther. 2000;294:800–809. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
