

Diagnosis and treatment trends in mucopolysaccharidosis I: findings from the MPS I Registry
- PMID: 22234477
- PMCID: PMC3357468
- DOI: 10.1007/s00431-011-1644-x
Diagnosis and treatment trends in mucopolysaccharidosis I: findings from the MPS I Registry
Abstract
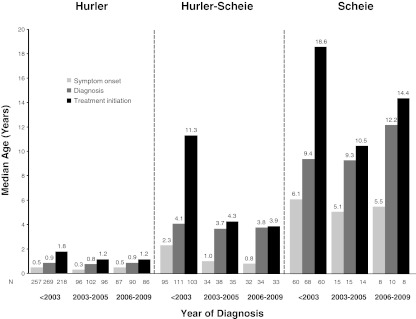
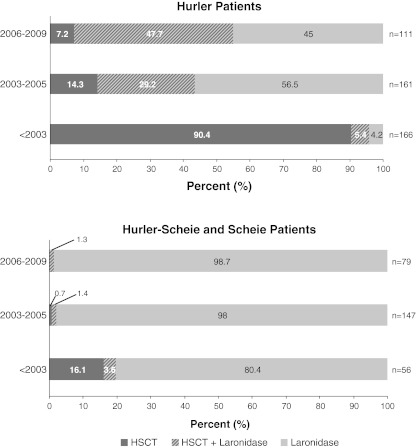
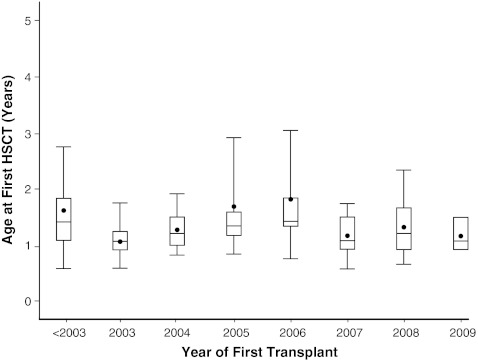
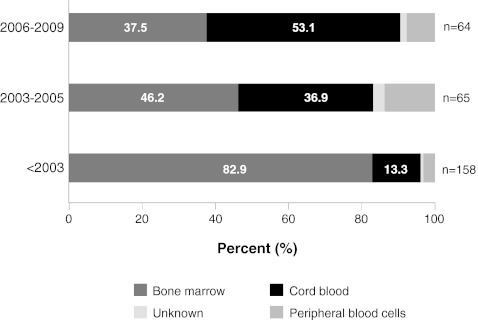
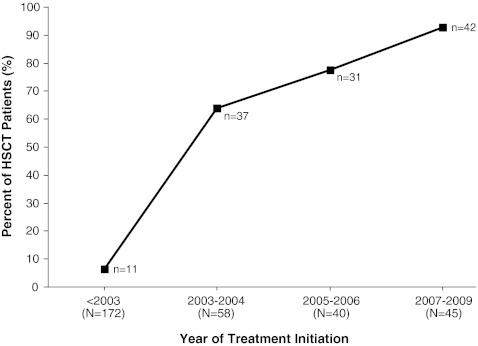
Our objective was to assess how the diagnosis and treatment of mucopolysaccharidosis I (MPS I) have changed over time. We used data from 891 patients in the MPS I Registry, an international observational database, to analyze ages at symptom onset, diagnosis, treatment initiation, and treatment allocation (hematopoietic stem cell transplantation, enzyme replacement therapy with laronidase, both, or neither) over time for all disease phenotypes (Hurler, Hurler-Scheie, and Scheie syndromes). The interval between diagnosis and treatment has become shorter since laronidase became available in 2003 (gap during 2006-2009: Hurler--0.2 year, Hurler-Scheie--0.5 year, Scheie--1.4 years). However, the age at diagnosis has not decreased for any MPS I phenotype over time, and the interval between symptom onset and treatment initiation remains substantial for both Hurler-Scheie and Scheie patients (gap during 2006-2009, 2.42 and 6.71 years, respectively). Among transplanted patients, an increasing proportion received hematopoietic stem cells from cord blood (34 out of 64 patients by 2009) and was also treated with laronidase (42 out of 45 patients by 2009).
Conclusions: Despite the availability of laronidase since 2003, the diagnosis of MPS I is still substantially delayed for patients with Hurler-Scheie and Scheie phenotypes, which can lead to a sub-optimal treatment outcome. Increased awareness of MPS I signs and symptoms by primary care providers and pediatric subspecialists is crucial to initiate early treatment and to improve the quality of life of MPS I patients.
Figures
References
-
- Arn P, Wraith J, Underhill L. Characterization of surgical procedures in patients with mucopolysaccharidosis type I: findings from the MPS I Registry. J Pediatr. 2009;154(859–864):e853. - PubMed
-
- Arn P, Whitley CB, Wraith JE, Underhill L, Rangachari L, Cox GF (2011) Postoperative mortality in patients with mucopolysaccharidosis I: findings from the MPS I Registry. J Pediatr Surg (in press) - PubMed
-
- Bodamer OA. Clinical characteristics of MPS I patients in the MPS I Registry. San Diego: The American Society of Human Genetics; 2007.
-
- Boelens JJ, Wynn RF, O’Meara A, Veys P, Bertrand Y, Souillet G, Wraith JE, Fischer A, Cavazzana-Calvo M, Sykora KW, Sedlacek P, Rovelli A, Uiterwaal CS, Wulffraat N. Outcomes of hematopoietic stem cell transplantation for Hurler’s syndrome in Europe: a risk factor analysis for graft failure. Bone Marrow Transplant. 2007;40:225–233. doi: 10.1038/sj.bmt.1705718. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
