

Virtual genomes in flux: an interplay of neutrality and adaptability explains genome expansion and streamlining
- PMID: 22234601
- PMCID: PMC3318439
- DOI: 10.1093/gbe/evr141
Virtual genomes in flux: an interplay of neutrality and adaptability explains genome expansion and streamlining
Abstract
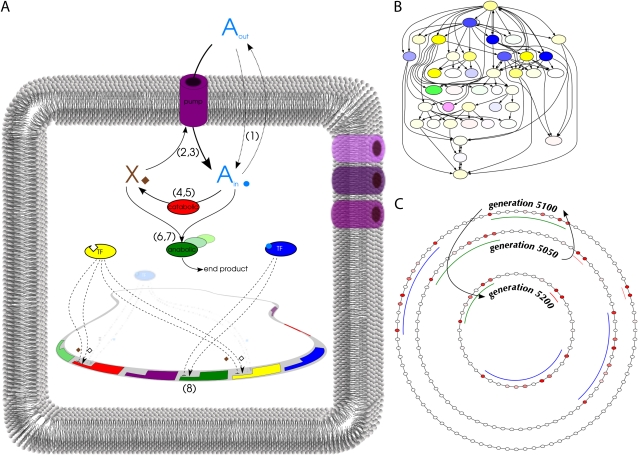
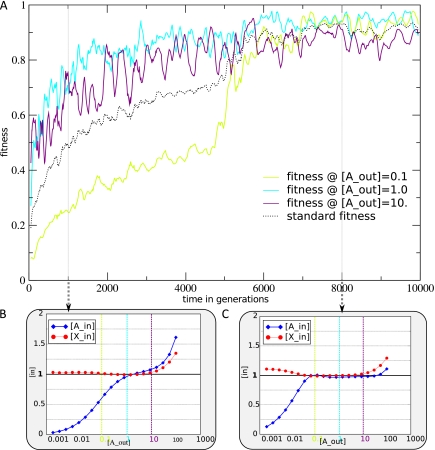
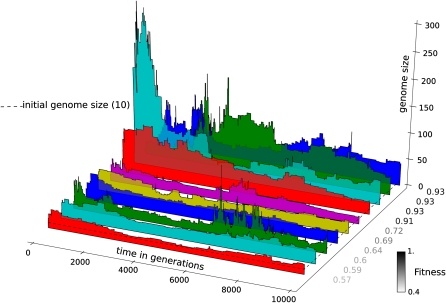
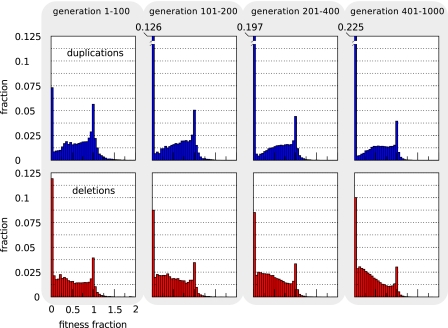
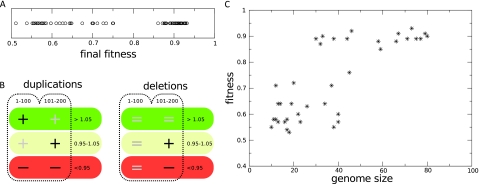
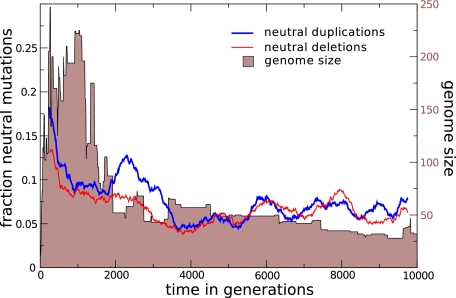
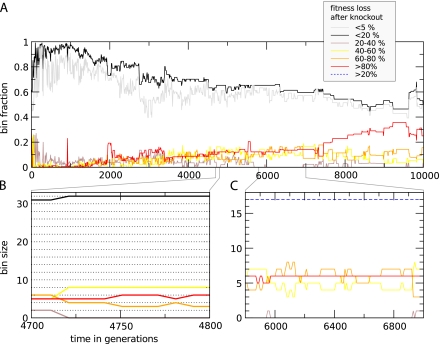
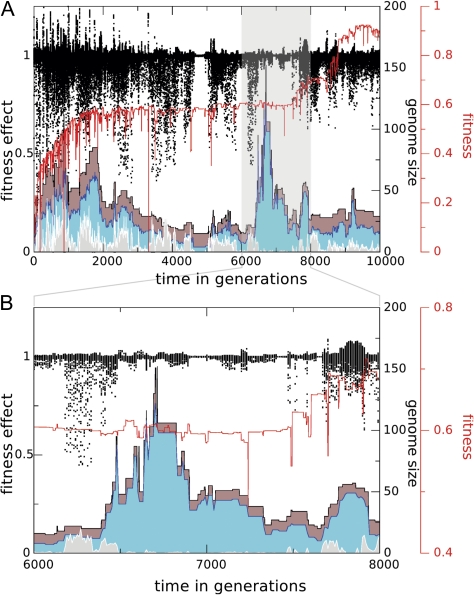
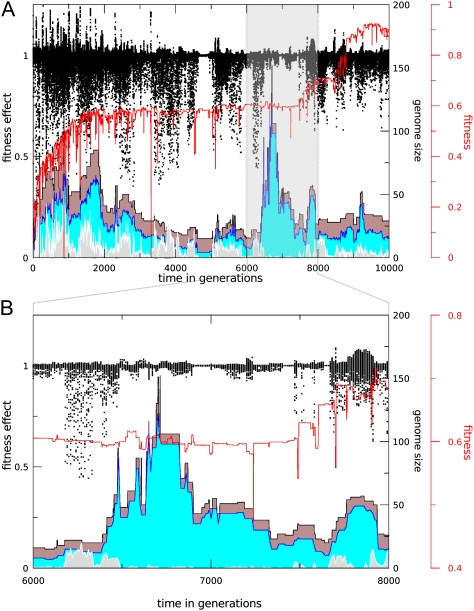
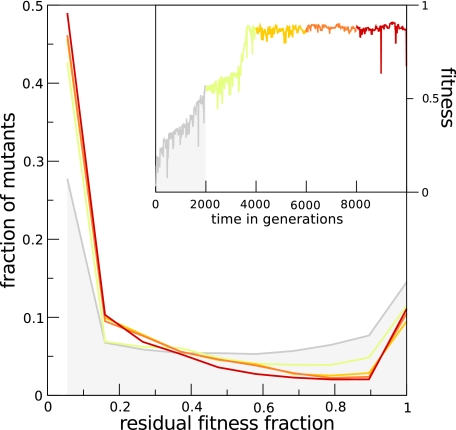
The picture that emerges from phylogenetic gene content reconstructions is that genomes evolve in a dynamic pattern of rapid expansion and gradual streamlining. Ancestral organisms have been estimated to possess remarkably rich gene complements, although gene loss is a driving force in subsequent lineage adaptation and diversification. Here, we study genome dynamics in a model of virtual cells evolving to maintain homeostasis. We observe a pattern of an initial rapid expansion of the genome and a prolonged phase of mutational load reduction. Generally, load reduction is achieved by the deletion of redundant genes, generating a streamlining pattern. Load reduction can also occur as a result of the generation of highly neutral genomic regions. These regions can expand and contract in a neutral fashion. Our study suggests that genome expansion and streamlining are generic patterns of evolving systems. We propose that the complex genotype to phenotype mapping in virtual cells as well as in their biological counterparts drives genome size dynamics, due to an emerging interplay between adaptation, neutrality, and evolvability.
Figures
References
-
- Aldana M, Balleza E, Kauffman S, Resendiz O. Robustness and evolvability in genetic regulatory networks. J Theor Biol. 2007;245:433–448. - PubMed
-
- Andersson DI, Hughes D. Gene amplification and adaptive evolution in bacteria. Annu Rev Genet. 2009;43(1):167–195. - PubMed
-
- Archibald JD. Divergence times of eutherian mammals. Science. 1999;285(5436):2031.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
