

Tumor Suppressor Function of CYLD in Nonmelanoma Skin Cancer
- PMID: 22235375
- PMCID: PMC3246786
- DOI: 10.1155/2011/614097
Tumor Suppressor Function of CYLD in Nonmelanoma Skin Cancer
Abstract
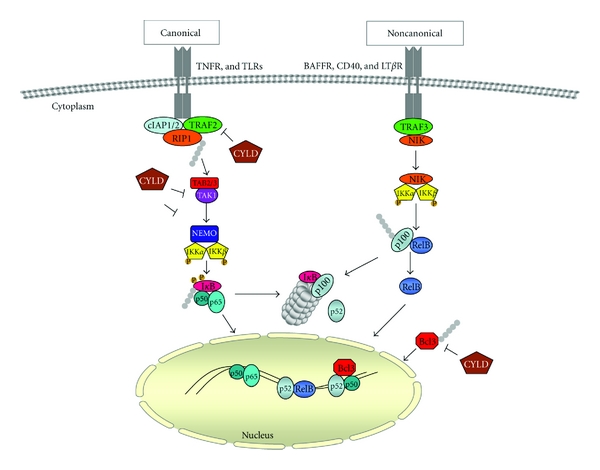
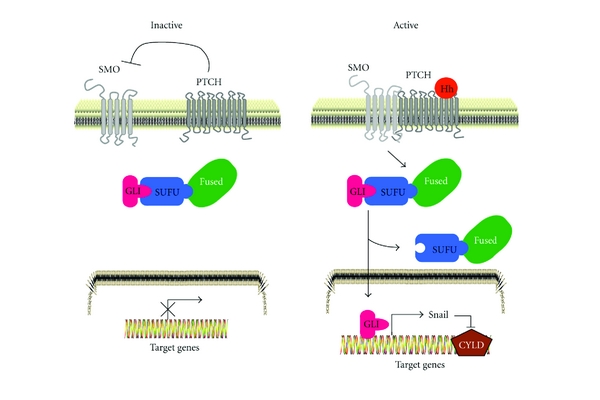
Ubiquitin and ubiquitin-related proteins posttranslationally modify substrates, and thereby alter the functions of their targets. The ubiquitination process is involved in various physiological responses, and dysregulation of components of the ubiquitin system has been linked to many diseases including skin cancer. The ubiquitin pathways activated among skin cancers are highly diverse and may reflect the various characteristics of the cancer type. Basal cell carcinoma and squamous cell carcinoma, the most common types of human skin cancer, are instances where the involvement of the deubiquitination enzyme CYLD has been recently highlighted. In basal cell carcinoma, the tumor suppressor protein CYLD is repressed at the transcriptional levels through hedgehog signaling pathway. Downregulation of CYLD in basal cell carcinoma was also shown to interfere with TrkC expression and signaling, thereby promoting cancer progression. By contrast, the level of CYLD is unchanged in squamous cell carcinoma, instead, catalytic inactivation of CYLD in the skin has been linked to the development of squamous cell carcinoma. This paper will focus on the current knowledge that links CYLD to nonmelanoma skin cancers and will explore recent insights regarding CYLD regulation of NF-κB and hedgehog signaling during the development and progression of these types of human tumors.
Figures


Similar articles
-
CYLD: a deubiquitination enzyme with multiple roles in cancer.Future Oncol. 2011 Feb;7(2):285-97. doi: 10.2217/fon.10.187. Future Oncol. 2011. PMID: 21345146 Review.
-
The E3 ubiquitin ligase MIB2 enhances inflammation by degrading the deubiquitinating enzyme CYLD.J Biol Chem. 2019 Sep 20;294(38):14135-14148. doi: 10.1074/jbc.RA119.010119. Epub 2019 Jul 31. J Biol Chem. 2019. PMID: 31366726 Free PMC article.
-
CYLD: a tumor suppressor deubiquitinase regulating NF-kappaB activation and diverse biological processes.Cell Death Differ. 2010 Jan;17(1):25-34. doi: 10.1038/cdd.2009.43. Cell Death Differ. 2010. PMID: 19373246 Free PMC article. Review.
-
Ubiquitin chain cleavage: CYLD at work.Trends Biochem Sci. 2010 Jul;35(7):392-9. doi: 10.1016/j.tibs.2010.02.007. Epub 2010 Mar 26. Trends Biochem Sci. 2010. PMID: 20347313 Review.
-
CYLD controls c-MYC expression through the JNK-dependent signaling pathway in hepatocellular carcinoma.Carcinogenesis. 2014 Feb;35(2):461-8. doi: 10.1093/carcin/bgt335. Epub 2013 Oct 8. Carcinogenesis. 2014. PMID: 24104553
Cited by
-
MiR-130b inhibits proliferation and induces apoptosis of gastric cancer cells via CYLD.Tumour Biol. 2016 Jun;37(6):7981-7. doi: 10.1007/s13277-015-4632-3. Epub 2015 Dec 28. Tumour Biol. 2016. PMID: 26711782
-
A computational approach for structural and functional analyses of disease-associated mutations in the human CYLD gene.Genomics Inform. 2024 May 31;22(1):4. doi: 10.1186/s44342-024-00007-2. Genomics Inform. 2024. PMID: 38907316 Free PMC article.
-
CYLD Alterations in the Tumorigenesis and Progression of Human Papillomavirus-Associated Head and Neck Cancers.Mol Cancer Res. 2021 Jan;19(1):14-24. doi: 10.1158/1541-7786.MCR-20-0565. Epub 2020 Sep 3. Mol Cancer Res. 2021. PMID: 32883697 Free PMC article. Review.
-
Premature aging and cancer development in transgenic mice lacking functional CYLD.Aging (Albany NY). 2019 Jan 10;11(1):127-159. doi: 10.18632/aging.101732. Aging (Albany NY). 2019. PMID: 30631004 Free PMC article.
-
miR-454 promotes survival and induces oxaliplatin resistance in gastric carcinoma cells by targeting CYLD.Exp Ther Med. 2020 Jun;19(6):3604-3610. doi: 10.3892/etm.2020.8655. Epub 2020 Apr 9. Exp Ther Med. 2020. PMID: 32346424 Free PMC article.
References
-
- Weissman AM. Themes and variations on ubiquitylation. Nature Reviews Molecular Cell Biology. 2001;2(3):169–178. - PubMed
-
- Pickart CM. Mechanisms underlying ubiquitination. Annual Review of Biochemistry. 2001;70:503–533. - PubMed
-
- Biggs PJ, Wooster R, Ford D, et al. Familial cylindromatosis (turban tumour syndrome) gene localised to chromosome 16q12-q13: evidence for its role as a tumour suppressor gene. Nature Genetics. 1995;11(4):441–443. - PubMed
-
- Bignell GR, Warren W, Seal S, et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nature Genetics. 2000;25(2):160–165. - PubMed
LinkOut - more resources
Full Text Sources
