

Cancer metabolism: current perspectives and future directions
- PMID: 22237205
- PMCID: PMC3270265
- DOI: 10.1038/cddis.2011.123
Cancer metabolism: current perspectives and future directions
Abstract
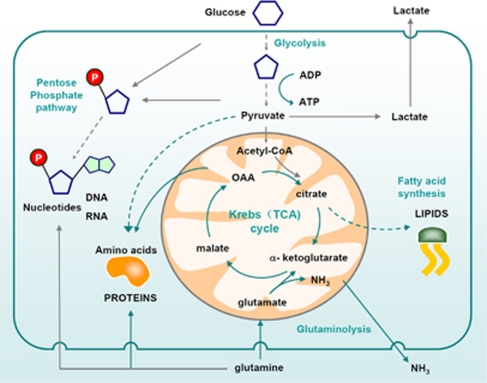
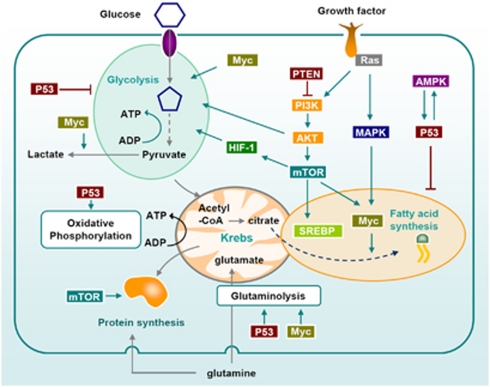
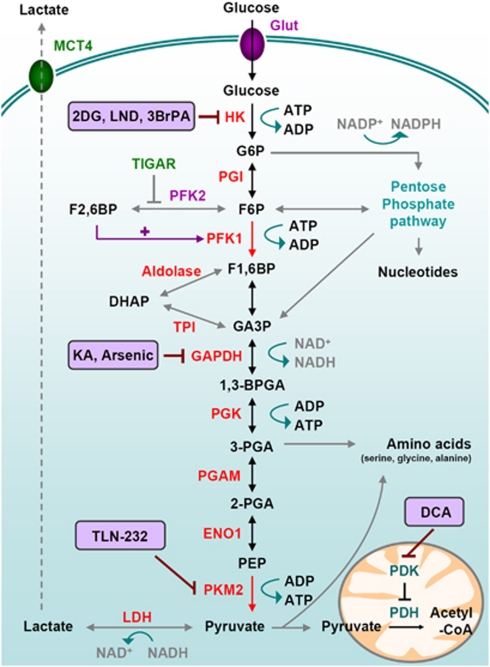
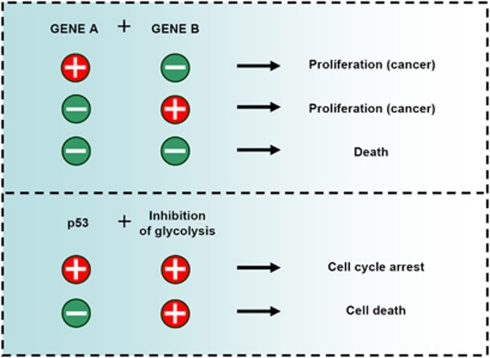
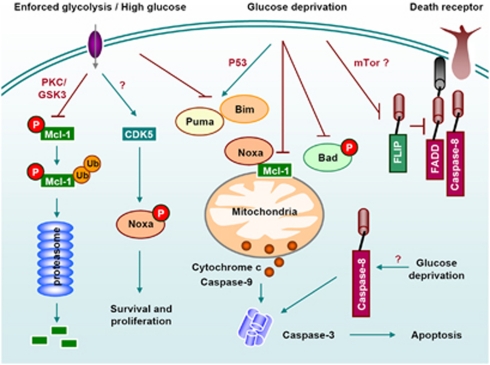
Cellular metabolism influences life and death decisions. An emerging theme in cancer biology is that metabolic regulation is intricately linked to cancer progression. In part, this is due to the fact that proliferation is tightly regulated by availability of nutrients. Mitogenic signals promote nutrient uptake and synthesis of DNA, RNA, proteins and lipids. Therefore, it seems straight-forward that oncogenes, that often promote proliferation, also promote metabolic changes. In this review we summarize our current understanding of how 'metabolic transformation' is linked to oncogenic transformation, and why inhibition of metabolism may prove a cancer's 'Achilles' heel'. On one hand, mutation of metabolic enzymes and metabolic stress sensors confers synthetic lethality with inhibitors of metabolism. On the other hand, hyperactivation of oncogenic pathways makes tumors more susceptible to metabolic inhibition. Conversely, an adequate nutrient supply and active metabolism regulates Bcl-2 family proteins and inhibits susceptibility to apoptosis. Here, we provide an overview of the metabolic pathways that represent anti-cancer targets and the cell death pathways engaged by metabolic inhibitors. Additionally, we will detail the similarities between metabolism of cancer cells and metabolism of proliferating cells.
Figures
References
-
- Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
