

Evidence for alkene cis-aminocupration, an aminooxygenation case study: kinetics, EPR spectroscopy, and DFT calculations
- PMID: 22237868
- PMCID: PMC3314301
- DOI: 10.1002/chem.201101703
Evidence for alkene cis-aminocupration, an aminooxygenation case study: kinetics, EPR spectroscopy, and DFT calculations
Abstract
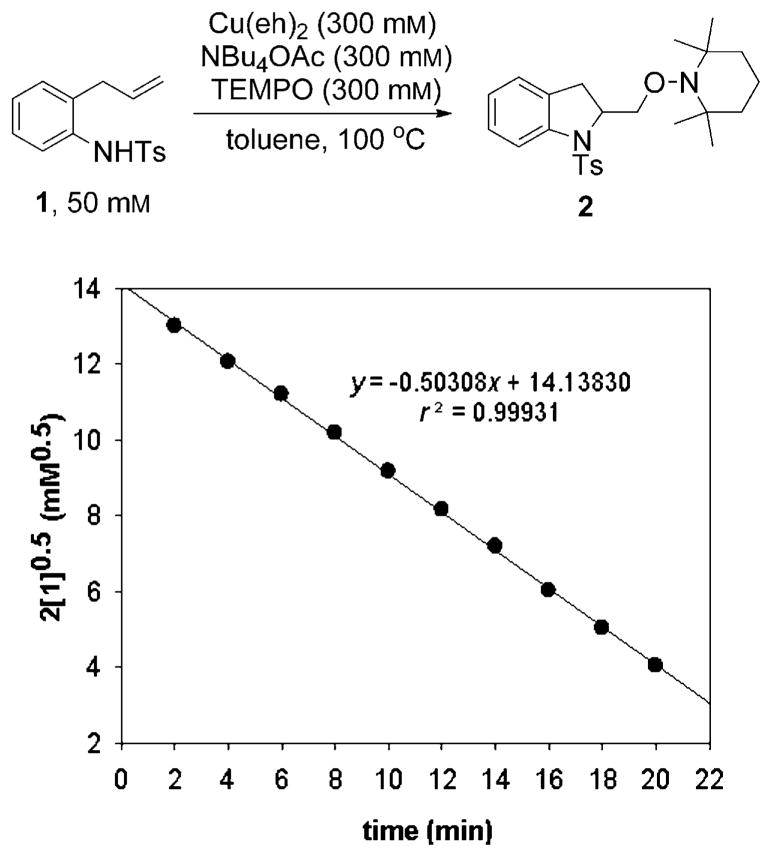
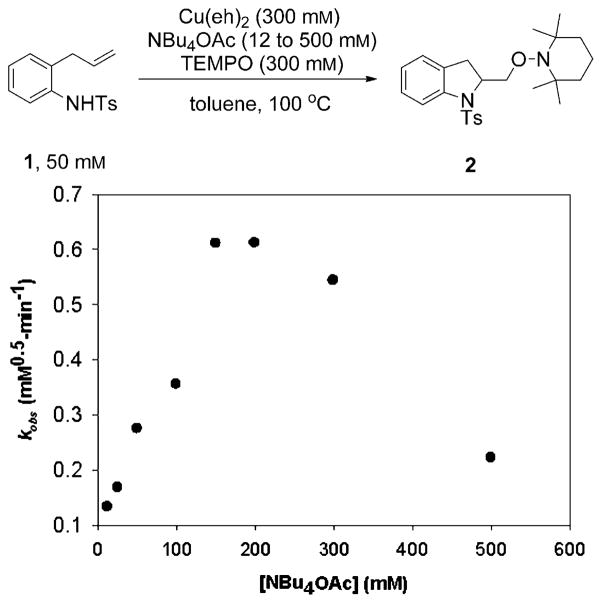
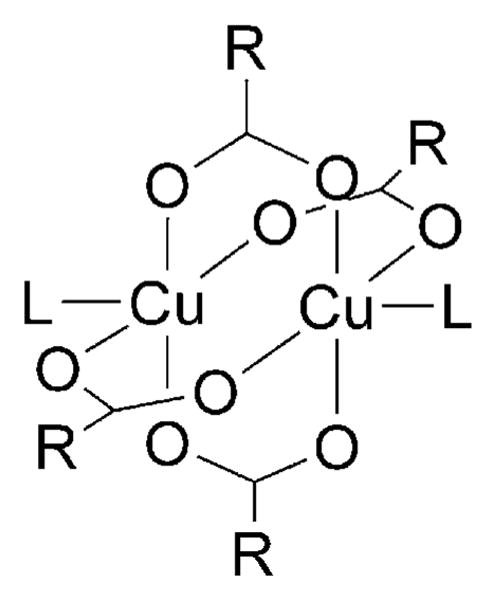
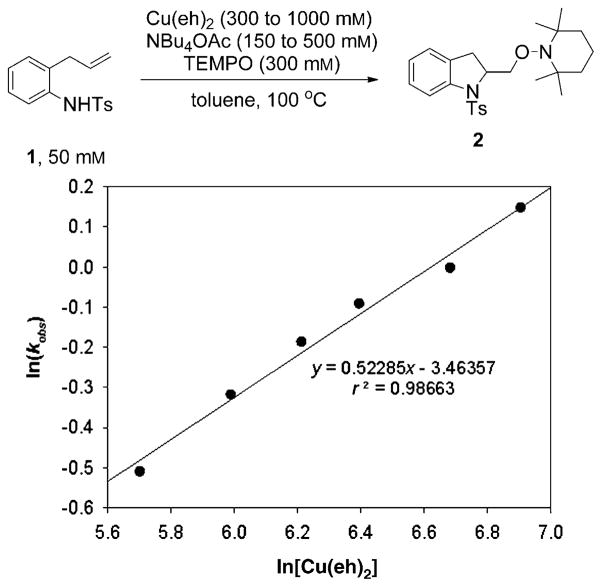
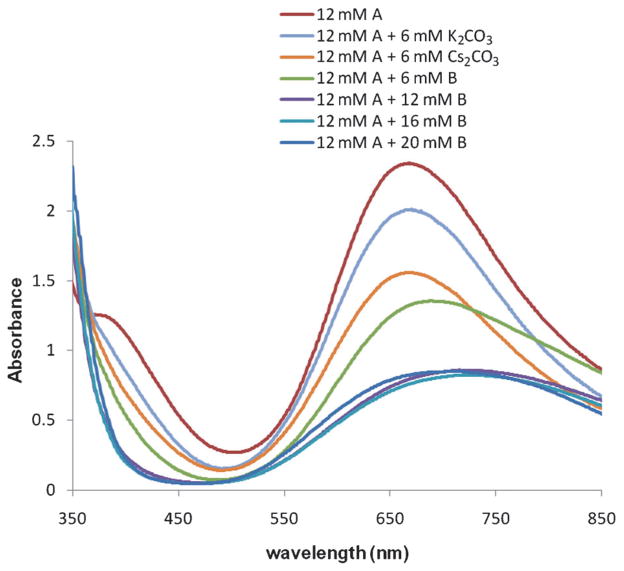
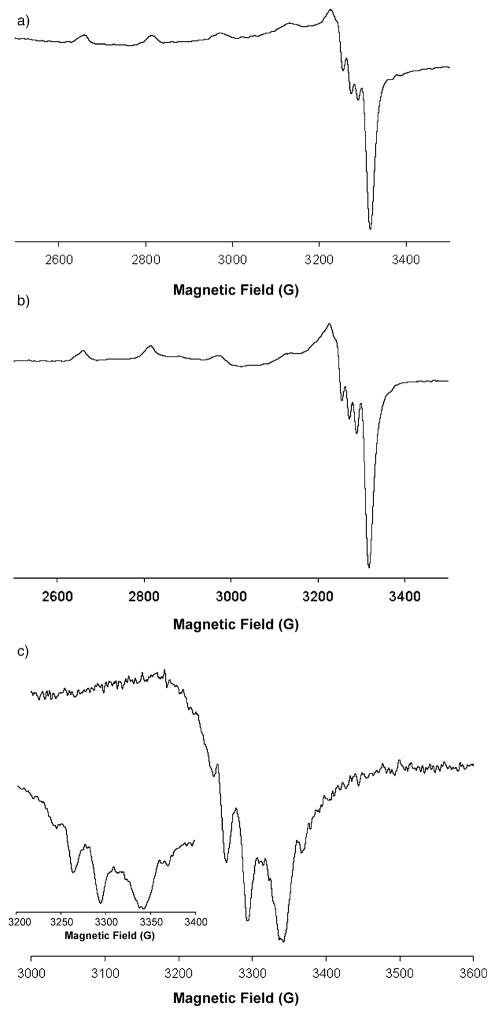
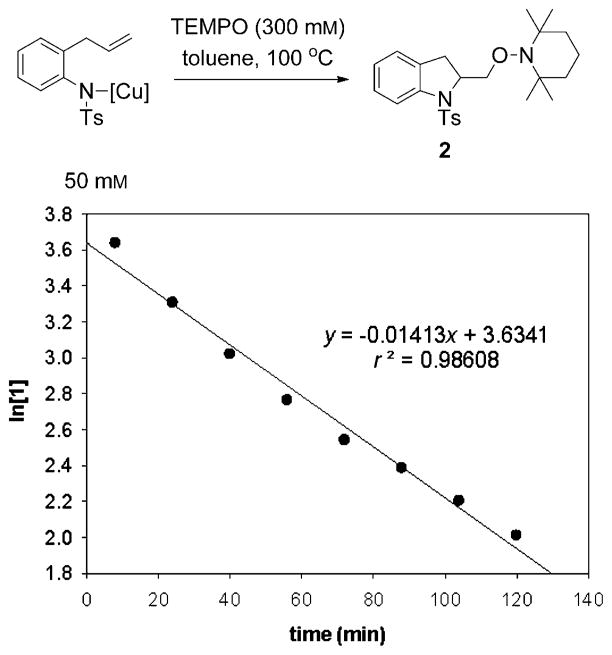
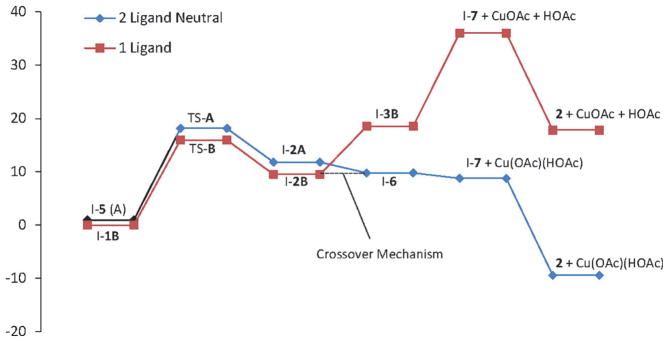
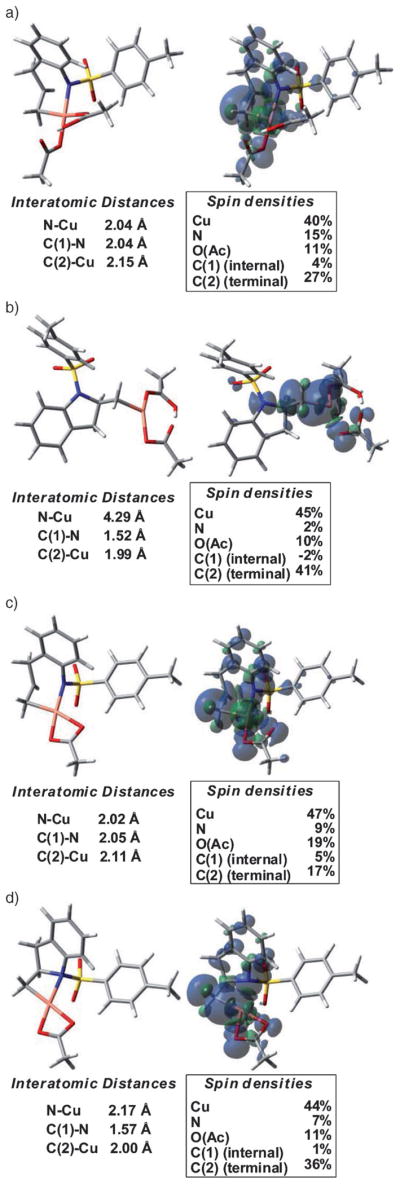
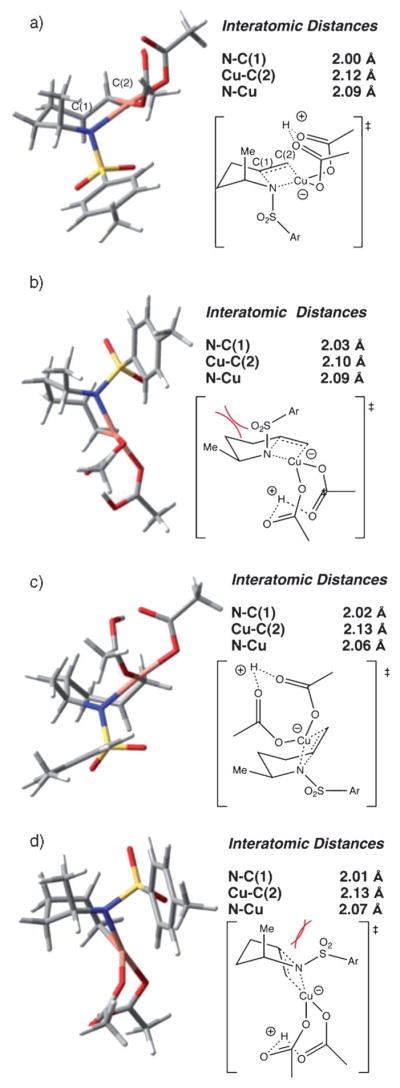
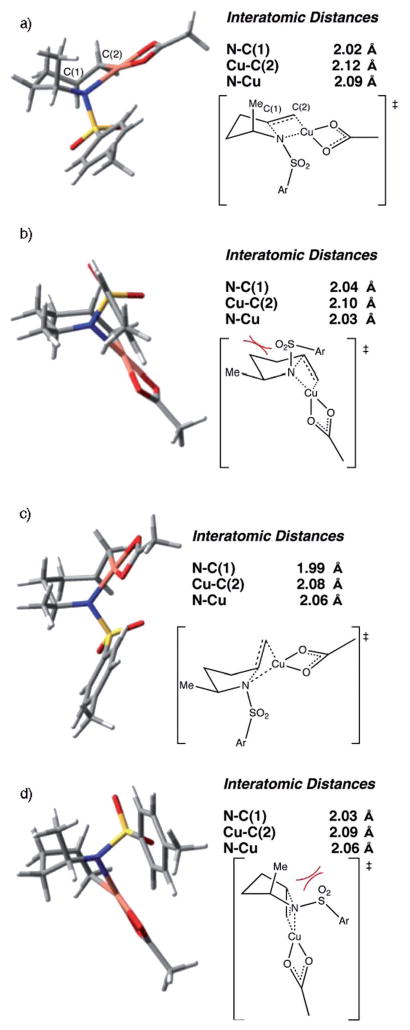
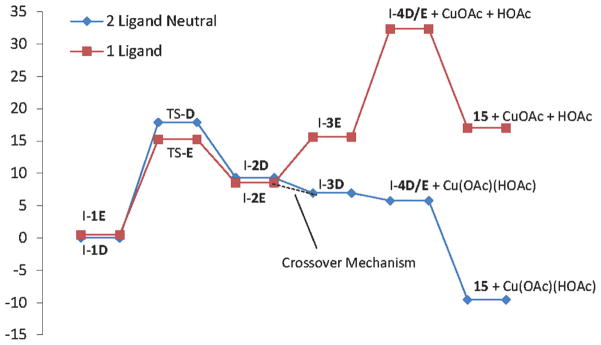
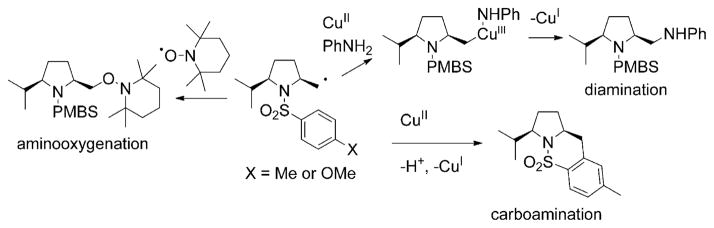
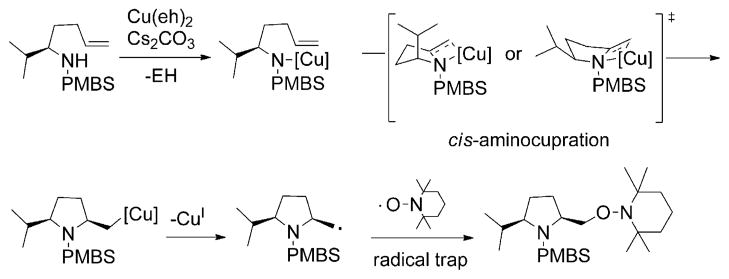
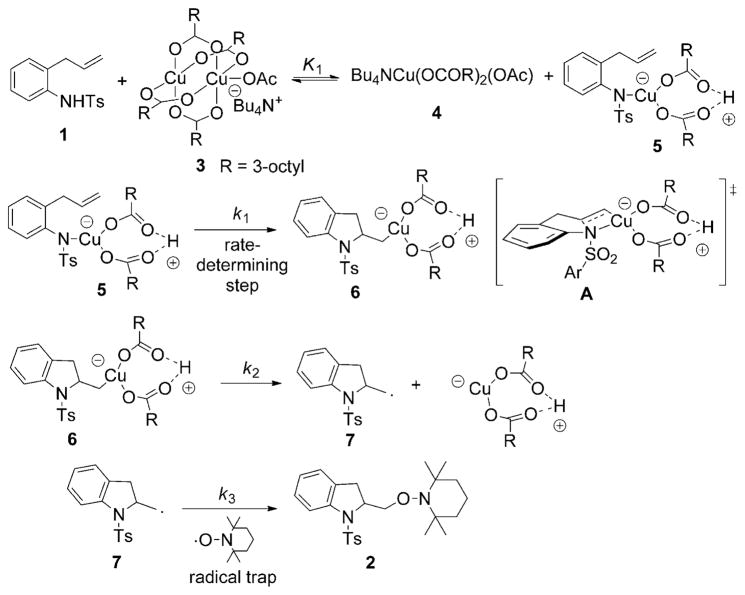
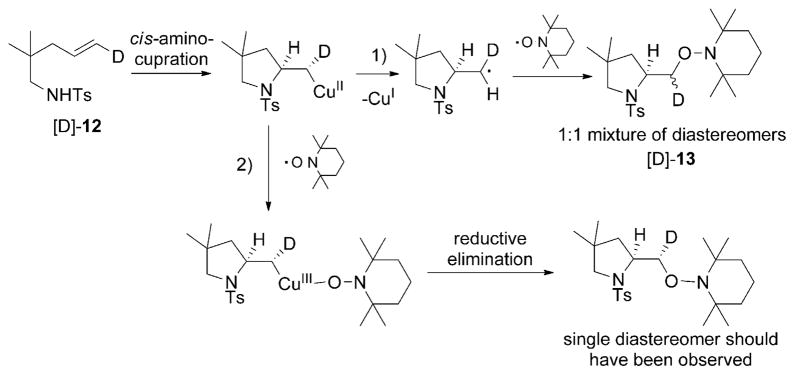
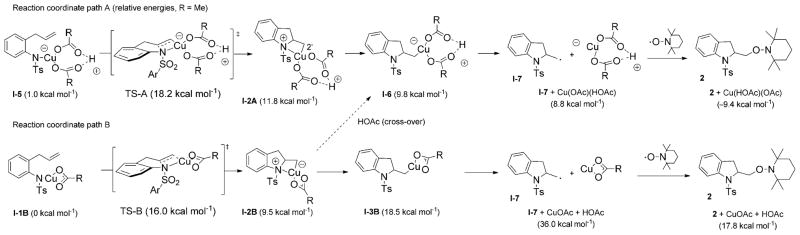
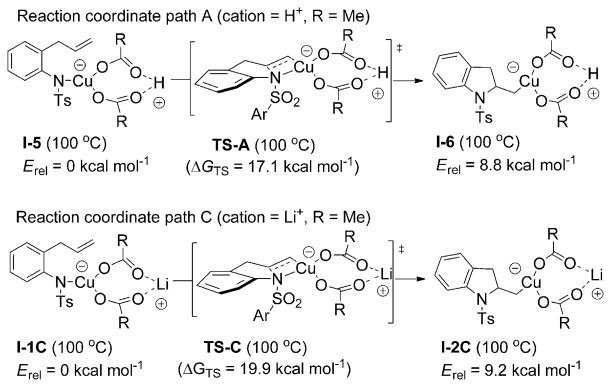
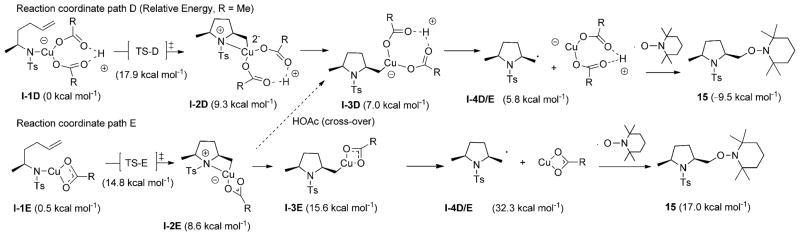
Alkene difunctionalization reactions are important in organic synthesis. We have recently shown that copper(II) complexes can promote and catalyze intramolecular alkene aminooxygenation, carboamination, and diamination reactions. In this contribution, we report a combined experimental and theoretical examination of the mechanism of the copper(II)-promoted olefin aminooxygenation reaction. Kinetics experiments revealed a mechanistic pathway involving an equilibrium reaction between a copper(II) carboxylate complex and the γ-alkenyl sulfonamide substrate and a rate-limiting intramolecular cis-addition of N-Cu across the olefin. Kinetic isotope effect studies support that the cis-aminocupration is the rate-determining step. UV/Vis spectra support a role for the base in the break-up of copper(II) carboxylate dimer to monomeric species. Electron paramagnetic resonance (EPR) spectra provide evidence for a kinetically competent N-Cu intermediate with a Cu(II) oxidation state. Due to the highly similar stereochemical and reactivity trends among the Cu(II)-promoted and catalyzed alkene difunctionalization reactions we have developed, the cis-aminocupration mechanism can reasonably be generalized across the reaction class. The methods and findings disclosed in this report should also prove valuable to the mechanism analysis and optimization of other copper(II) carboxylate promoted reactions, especially those that take place in aprotic organic solvents.
Copyright © 2012 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
