

Estimating abundances of retroviral insertion sites from DNA fragment length data
- PMID: 22238265
- PMCID: PMC3307109
- DOI: 10.1093/bioinformatics/bts004
Estimating abundances of retroviral insertion sites from DNA fragment length data
Abstract
Motivation: The relative abundance of retroviral insertions in a host genome is important in understanding the persistence and pathogenesis of both natural retroviral infections and retroviral gene therapy vectors. It could be estimated from a sample of cells if only the host genomic sites of retroviral insertions could be directly counted. When host genomic DNA is randomly broken via sonication and then amplified, amplicons of varying lengths are produced. The number of unique lengths of amplicons of an insertion site tends to increase according to its abundance, providing a basis for estimating relative abundance. However, as abundance increases amplicons of the same length arise by chance leading to a non-linear relation between the number of unique lengths and relative abundance. The difficulty in calibrating this relation is compounded by sample-specific variations in the relative frequencies of clones of each length.
Results: A likelihood function is proposed for the discrete lengths observed in each of a collection of insertion sites and is maximized with a hybrid expectation-maximization algorithm. Patient data illustrate the method and simulations show that relative abundance can be estimated with little bias, but that variation in highly abundant sites can be large. In replicated patient samples, variation exceeds what the model implies-requiring adjustment as in Efron (2004) or using jackknife standard errors. Consequently, it is advantageous to collect replicate samples to strengthen inferences about relative abundance.
Figures

 versus Length. Estimates are provided for the replicates of sample I1 (solid lines) and sample B2 (dashed lines). The insert (dotted box) shows the corresponding calibration curves and an empirical calibration curve (thick line—see text).
versus Length. Estimates are provided for the replicates of sample I1 (solid lines) and sample B2 (dashed lines). The insert (dotted box) shows the corresponding calibration curves and an empirical calibration curve (thick line—see text).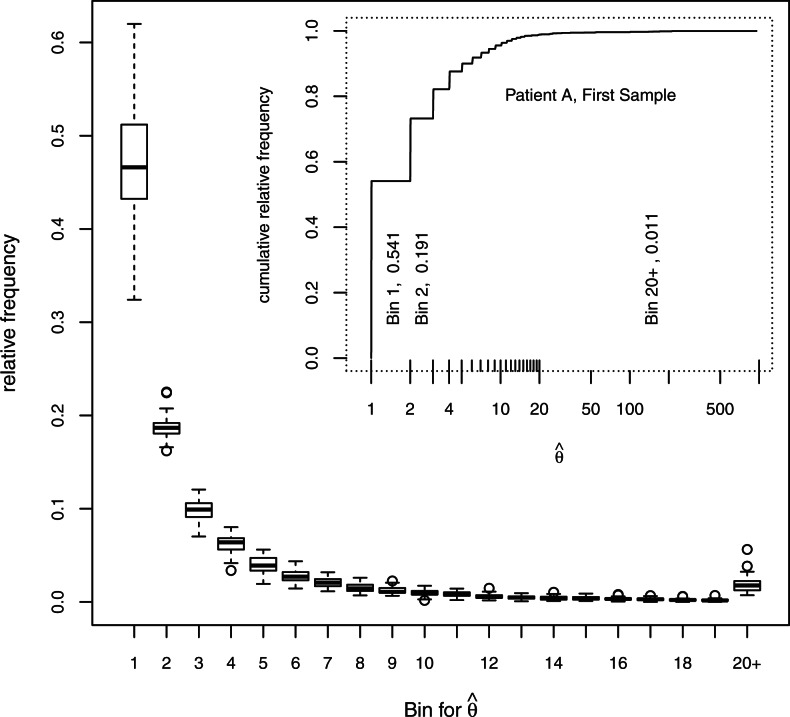
 for 33 samples. The box covers the first through third quartiles of the data, the central line of each box shows the median, the whiskers extend to the closer of the extreme or to 1.5 times the height of the box away from the box, and circles show points, if any, that lie beyond the whiskers.
for 33 samples. The box covers the first through third quartiles of the data, the central line of each box shows the median, the whiskers extend to the closer of the extreme or to 1.5 times the height of the box away from the box, and circles show points, if any, that lie beyond the whiskers.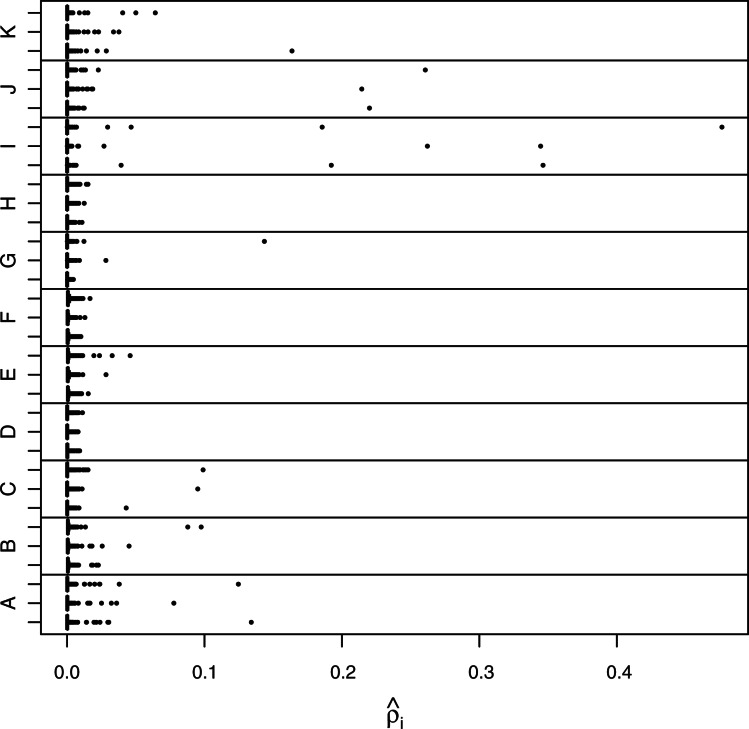

References
-
- Baker S. The multinomial-poisson transformation. Statistician. 1994;43:495–504.
-
- Chao A. Estimating the population size for capture-recapture data with unequal catchability. Biometrics. 1987;43:783–791. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
