

The viral protein Tat can inhibit the establishment of HIV-1 latency
- PMID: 22238306
- PMCID: PMC3302319
- DOI: 10.1128/JVI.06648-11
The viral protein Tat can inhibit the establishment of HIV-1 latency
Abstract
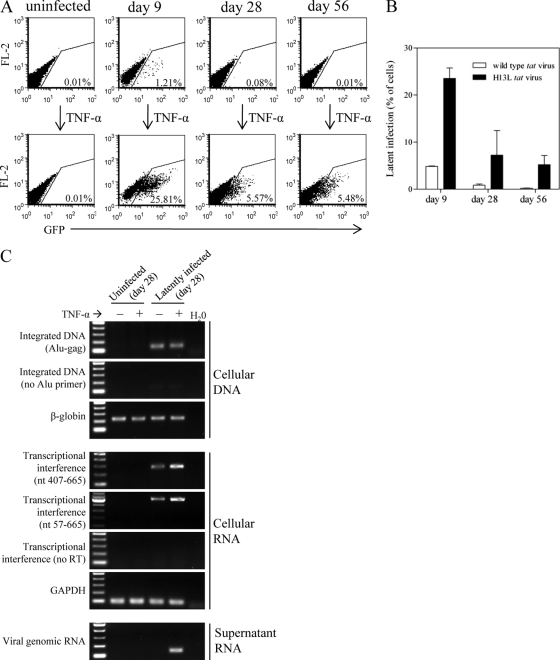
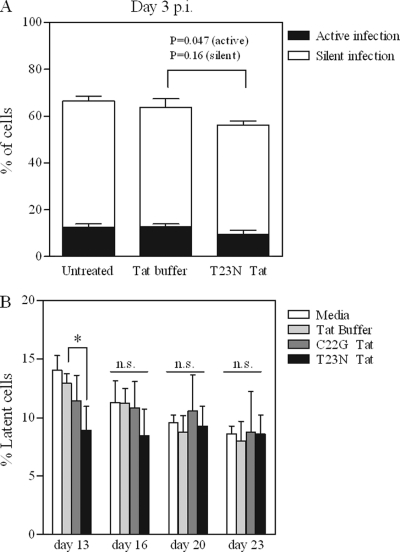
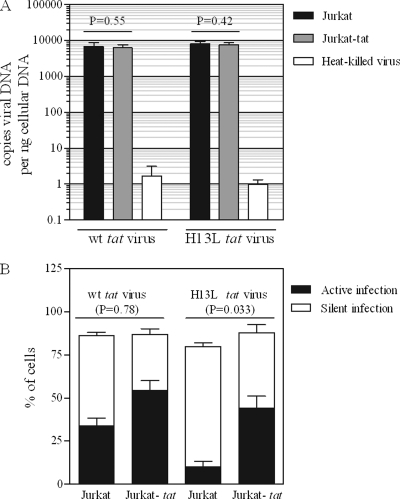
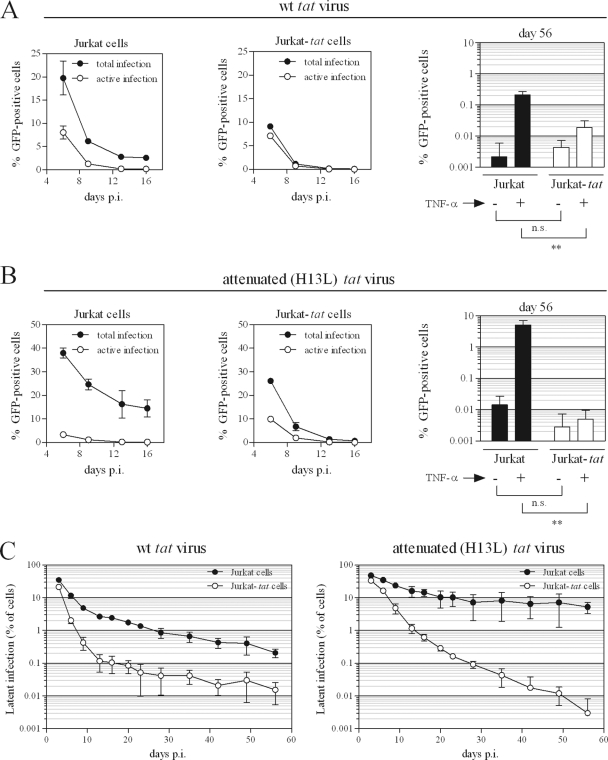
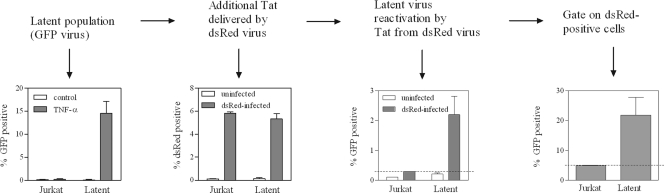
The establishment of HIV-1 latency can result from limiting levels of transcription initiation or elongation factors, restrictive chromatin modifications, transcriptional interference, and insufficient Tat activity. Since the viral protein Tat can counteract many of these factors, we hypothesized that the presence of exogenous Tat during infection might inhibit the establishment of latency. This was explored using a Jurkat model of latency establishment and reactivation. PCR and reverse transcriptase PCR (RT-PCR) confirmed the latent state in this model and showed evidence of transcriptional interference. To address our hypothesis, cells undergoing infection were first exposed to either purified recombinant Tat or a transactivation-negative mutant. Only the former resulted in a modest inhibition of the establishment of latency. Next, Jurkat cells stably expressing intracellular Tat were used in our latency model to avoid limitations of Tat delivery. Experiments confirmed that intracellular Tat expression did not affect the susceptibility of these cells to viral infection. Eight weeks after infection, Jurkat cells expressing Tat harbored up to 1,700-fold fewer (P < 0.01) latent viruses than Jurkat cells that did not express Tat. Additionally, Tat delivered by a second virus was sufficient to reactivate most of the latent population. Our results suggest that inhibition of the establishment of latent infection is theoretically possible. In a hypothetical scenario of therapy that induces viral gene expression during acute infection, activation of viruses which would otherwise have entered latency could occur while concurrent highly active antiretroviral therapy (HAART) would prevent further viral spread, potentially decreasing the size of the established latent reservoir.
Figures

Similar articles
-
A Stronger Transcription Regulatory Circuit of HIV-1C Drives the Rapid Establishment of Latency with Implications for the Direct Involvement of Tat.J Virol. 2020 Sep 15;94(19):e00503-20. doi: 10.1128/JVI.00503-20. Print 2020 Sep 15. J Virol. 2020. PMID: 32669338 Free PMC article.
-
Variation in HIV-1 Tat activity is a key determinant in the establishment of latent infection.JCI Insight. 2024 Dec 5;10(2):e184711. doi: 10.1172/jci.insight.184711. JCI Insight. 2024. PMID: 39636695 Free PMC article.
-
BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism.Cell Cycle. 2013 Feb 1;12(3):452-62. doi: 10.4161/cc.23309. Epub 2012 Feb 1. Cell Cycle. 2013. PMID: 23255218 Free PMC article.
-
Lost in transcription: molecular mechanisms that control HIV latency.Viruses. 2013 Mar 21;5(3):902-27. doi: 10.3390/v5030902. Viruses. 2013. PMID: 23518577 Free PMC article. Review.
-
The Molecular Basis for Human Immunodeficiency Virus Latency.Annu Rev Virol. 2017 Sep 29;4(1):261-285. doi: 10.1146/annurev-virology-101416-041646. Epub 2017 Jul 17. Annu Rev Virol. 2017. PMID: 28715973 Review.
Cited by
-
HIV-1 Tat amino acid residues that influence Tat-TAR binding affinity: a scoping review.BMC Infect Dis. 2023 Mar 17;23(1):164. doi: 10.1186/s12879-023-08123-0. BMC Infect Dis. 2023. PMID: 36932337 Free PMC article.
-
Antiretroviral therapy initiated during acute infection in women with HIV-1 clade C reduces anti-Tat antibody production and lowers CD8+ T cell activation.Front Immunol. 2025 Jun 18;16:1564960. doi: 10.3389/fimmu.2025.1564960. eCollection 2025. Front Immunol. 2025. PMID: 40607402 Free PMC article.
-
The Tat Inhibitor Didehydro-Cortistatin A Prevents HIV-1 Reactivation from Latency.mBio. 2015 Jul 7;6(4):e00465. doi: 10.1128/mBio.00465-15. mBio. 2015. PMID: 26152583 Free PMC article.
-
HIV-1 transcriptional modulation: novel host factors and prospective therapeutic strategies.Curr Opin HIV AIDS. 2023 Sep 1;18(5):264-272. doi: 10.1097/COH.0000000000000808. Epub 2023 Jul 17. Curr Opin HIV AIDS. 2023. PMID: 37535041 Free PMC article. Review.
-
Tat inhibition by didehydro-Cortistatin A promotes heterochromatin formation at the HIV-1 long terminal repeat.Epigenetics Chromatin. 2019 Apr 16;12(1):23. doi: 10.1186/s13072-019-0267-8. Epigenetics Chromatin. 2019. PMID: 30992052 Free PMC article.
References
-
- Benkirane M, et al. 1998. Activation of integrated provirus requires histone acetyltransferase. p300 and P/CAF are coactivators for HIV-1 Tat. J. Biol. Chem. 273:24898–24905 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
