

Rapid gene-based SNP and haplotype marker development in non-model eukaryotes using 3'UTR sequencing
- PMID: 22239826
- PMCID: PMC3293726
- DOI: 10.1186/1471-2164-13-18
Rapid gene-based SNP and haplotype marker development in non-model eukaryotes using 3'UTR sequencing
Abstract
Background: Sweet cherry (Prunus avium L.), a non-model crop with narrow genetic diversity, is an important member of sub-family Amygdoloideae within Rosaceae. Compared to other important members like peach and apple, sweet cherry lacks in genetic and genomic information, impeding understanding of important biological processes and development of efficient breeding approaches. Availability of single nucleotide polymorphism (SNP)-based molecular markers can greatly benefit breeding efforts in such non-model species. RNA-seq approaches employing second generation sequencing platforms offer a unique avenue to rapidly identify gene-based SNPs. Additionally, haplotype markers can be rapidly generated from transcript-based SNPs since they have been found to be extremely utile in identification of genetic variants related to health, disease and response to environment as highlighted by the human HapMap project.
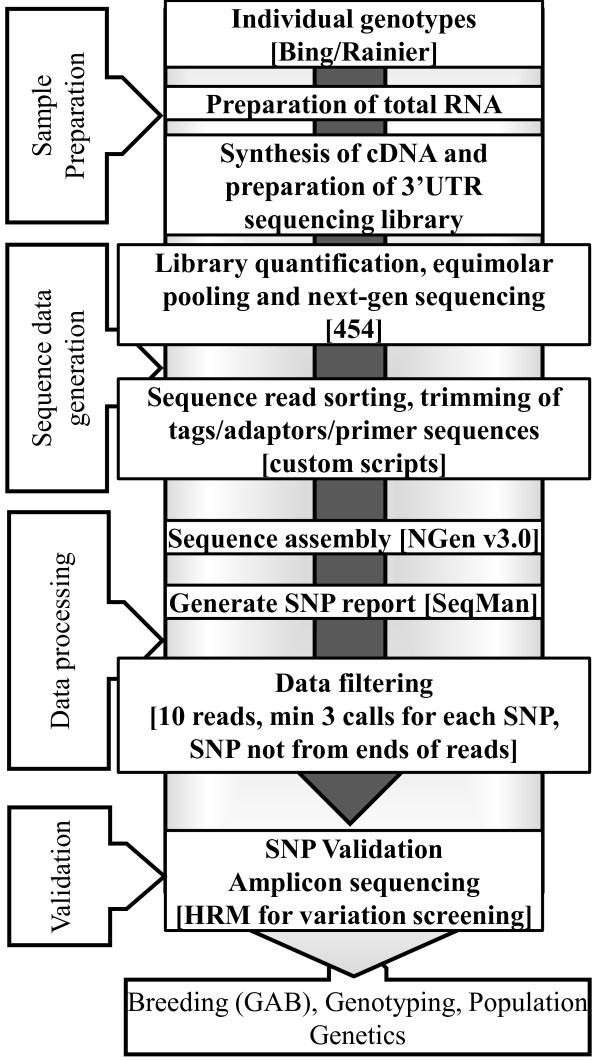
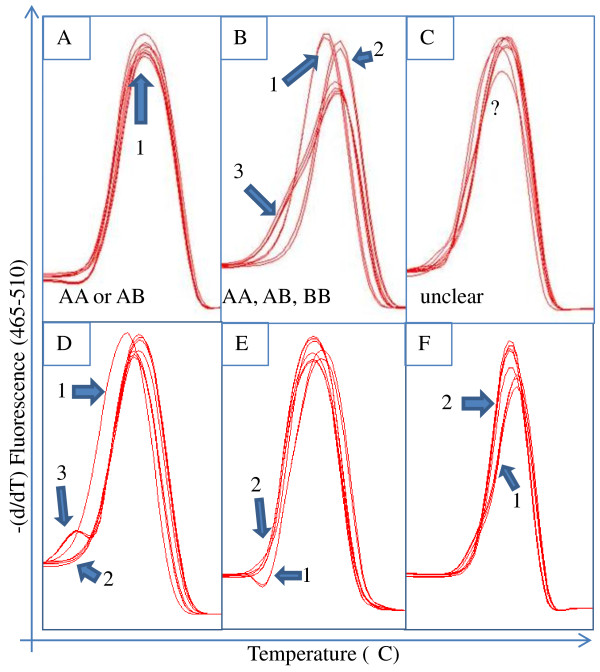
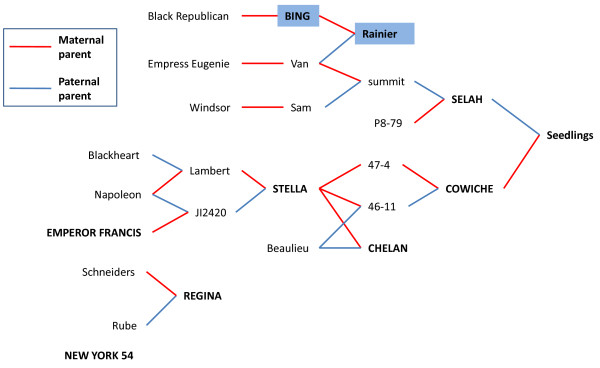
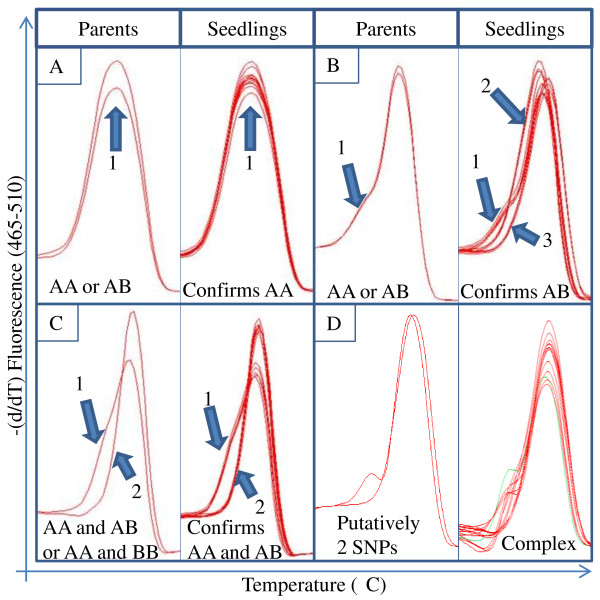
Results: RNA-seq was performed on two sweet cherry cultivars, Bing and Rainier using a 3' untranslated region (UTR) sequencing method yielding 43,396 assembled contigs. In order to test our approach of rapid identification of SNPs without any reference genome information, over 25% (10,100) of the contigs were screened for the SNPs. A total of 207 contigs from this set were identified to contain high quality SNPs. A set of 223 primer pairs were designed to amplify SNP containing regions from these contigs and high resolution melting (HRM) analysis was performed with eight important parental sweet cherry cultivars. Six of the parent cultivars were distantly related to Bing and Rainier, the cultivars used for initial SNP discovery. Further, HRM analysis was also performed on 13 seedlings derived from a cross between two of the parents. Our analysis resulted in the identification of 84 (38.7%) primer sets that demonstrated variation among the tested germplasm. Reassembly of the raw 3'UTR sequences using upgraded transcriptome assembly software yielded 34,620 contigs containing 2243 putative SNPs in 887 contigs after stringent filtering. Contigs with multiple SNPs were visually parsed to identify 685 putative haplotypes at 335 loci in 301 contigs.
Conclusions: This approach, which leverages the advantages of RNA-seq approaches, enabled rapid generation of gene-linked SNP and haplotype markers. The general approach presented in this study can be easily applied to other non-model eukaryotes irrespective of the ploidy level to identify gene-linked polymorphisms that are expected to facilitate efficient Gene Assisted Breeding (GAB), genotyping and population genetics studies. The identified SNP haplotypes reveal some of the allelic differences in the two sweet cherry cultivars analyzed. The identification of these SNP and haplotype markers is expected to significantly improve the genomic resources for sweet cherry and facilitate efficient GAB in this non-model crop.
Figures

References
-
- Arumuganathan K, Earle E. Nuclear DNA content of some important plant species. Plant Molecular Biology Reporter. 1991;9(3):208–218. doi: 10.1007/BF02672069. - DOI
-
- Peach Genome v1.0, International Peach Genome Initiative. http://www.phytozome.net/peach.php#A
-
- Li S, Yin T, Wang M, Tuskan G. Characterization of microsatellites in the coding regions of the Populus genome. Molecular Breeding. 2011;27(1):59–66. doi: 10.1007/s11032-010-9413-5. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
