

Enhancement of lung tumorigenesis in a Gprc5a Knockout mouse by chronic extrinsic airway inflammation
- PMID: 22239913
- PMCID: PMC3281775
- DOI: 10.1186/1476-4598-11-4
Enhancement of lung tumorigenesis in a Gprc5a Knockout mouse by chronic extrinsic airway inflammation
Abstract
Background: Although cigarette smoking is the principal cause of lung carcinogenesis, chronic obstructive pulmonary disease (COPD), an inflammatory disease of the lung, has been identified as an independent risk factor for lung cancer. Bacterial colonization, particularly with non-typeable Haemophilus influenzae (NTHi), has been implicated as a cause of airway inflammation in COPD besides cigarette smoke. Accordingly, we hypothesized that lung cancer promotion may occur in a chronic inflammatory environment in the absence of concurrent carcinogen exposure.
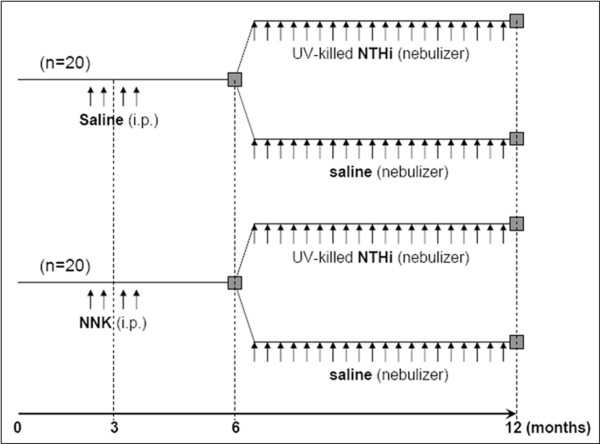
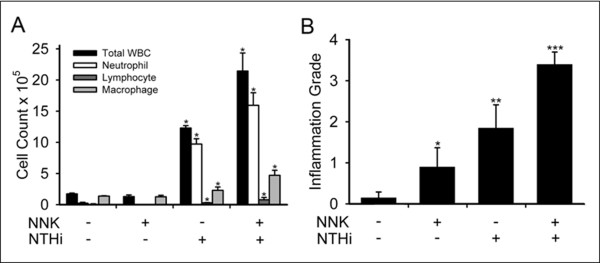
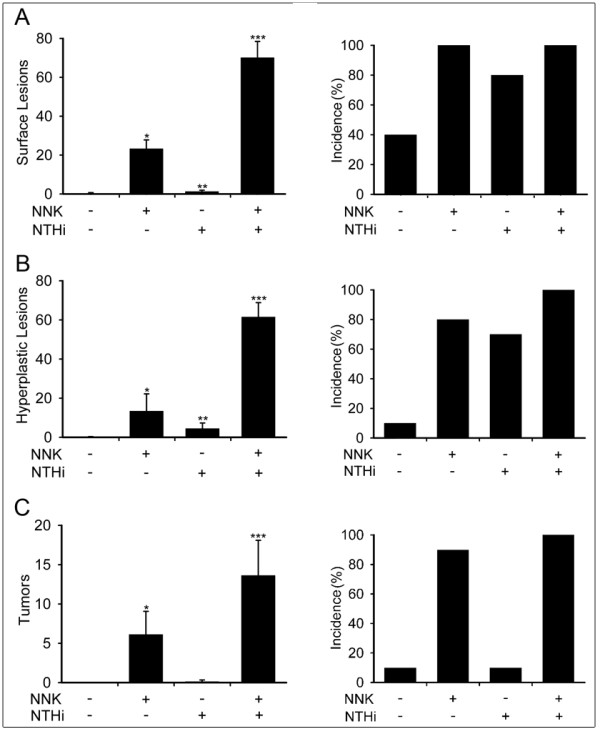
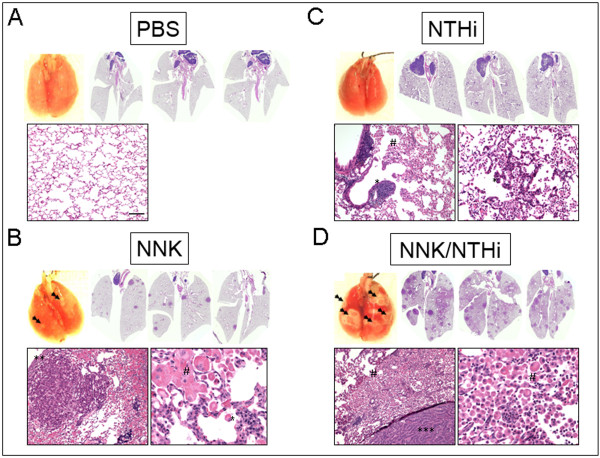
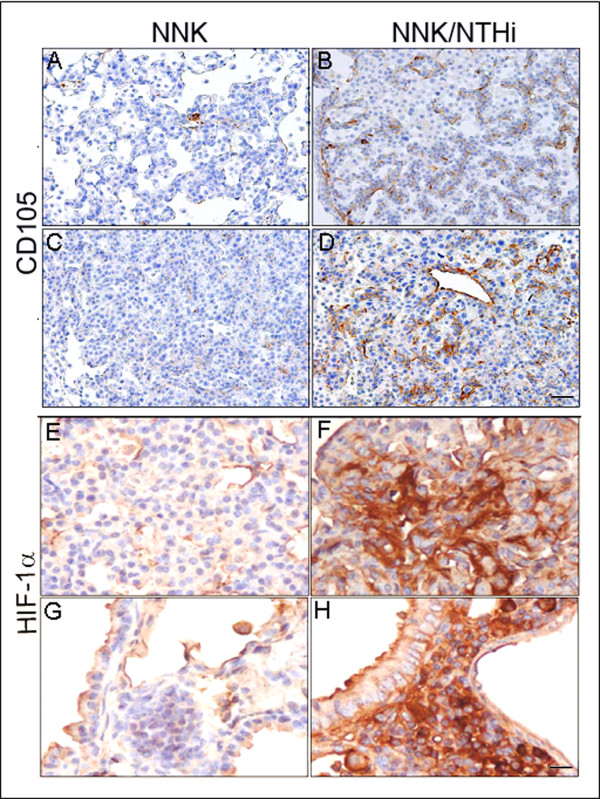
Results: Herein, we investigated the effects of bacterial-induced COPD-like inflammation and tobacco carcinogen-enhanced tumorigenesis/inflammation in the retinoic acid inducible G protein coupled receptor knock out mouse model (Gprc5a-/- mouse) characterized by late-onset, low multiplicity tumor formation. Three-month-old Gprc5a-/- mice received 4 intraperitoneal injections of the tobacco-specific carcinogen, NNK, followed by weekly exposure to aerosolized NTHi lysate for 6 months. The numbers of inflammatory cells in the lungs and levels of several inflammatory mediators were increased in Gprc5a-/- mice treated with NTHi alone, and even more so in mice pretreated with NNK followed by NTHi. The incidence of spontaneous lung lesions in the Gprc5a-/- mice was low, but NTHi exposure led to enhanced development of hyperplastic lesions. Gprc5a-/- mice exposed to NNK alone developed multiple lung tumors, while NTHi exposure increased the number of hyperplastic foci 6-fold and the tumor multiplicity 2-fold. This was associated with increased microvessel density and HIF-1α expression.
Conclusion: We conclude that chronic extrinsic lung inflammation induced by bacteria alone or in combination with NNK enhances lung tumorigenesis in Gprc5a-/- mice.
Figures
References
-
- Stellman SD, Takezaki T, Wang L, Chen Y, Citron ML, Djordjevic MV, Harlap S, Muscat JE, Neugut AI, Wynder EL. et al.Smoking and lung cancer risk in American and Japanese men: an international case-control study. Cancer Epidemiol Biomarkers Prev. 2001;10:1193–1199. - PubMed
-
- Schottenfield D. In: Lung cancer: Principles and Practice. Pass HIMJBJDHTATMJD, editor. Philadelphia: Lippincott, Williams and Wilkins; 2000. Etiology and epidemiology of lung cancer; pp. 367–388.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
