

Diverse functional properties of Wilson disease ATP7B variants
- PMID: 22240481
- PMCID: PMC3461965
- DOI: 10.1053/j.gastro.2011.12.048
Diverse functional properties of Wilson disease ATP7B variants
Abstract
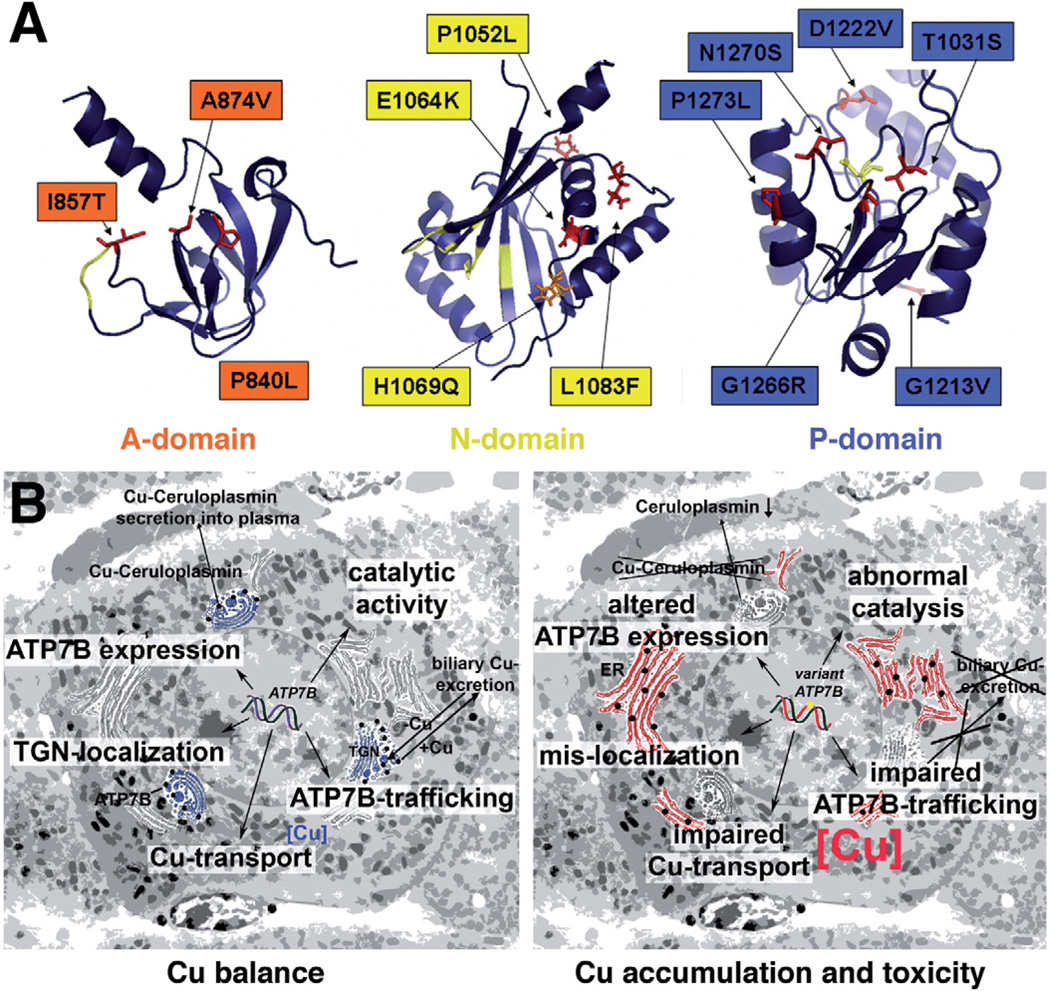
Background & aims: Wilson disease is a severe disorder of copper metabolism caused by mutations in ATP7B, which encodes a copper-transporting adenosine triphosphatase. The disease presents with a variable phenotype that complicates the diagnostic process and treatment. Little is known about the mechanisms that contribute to the different phenotypes of the disease.
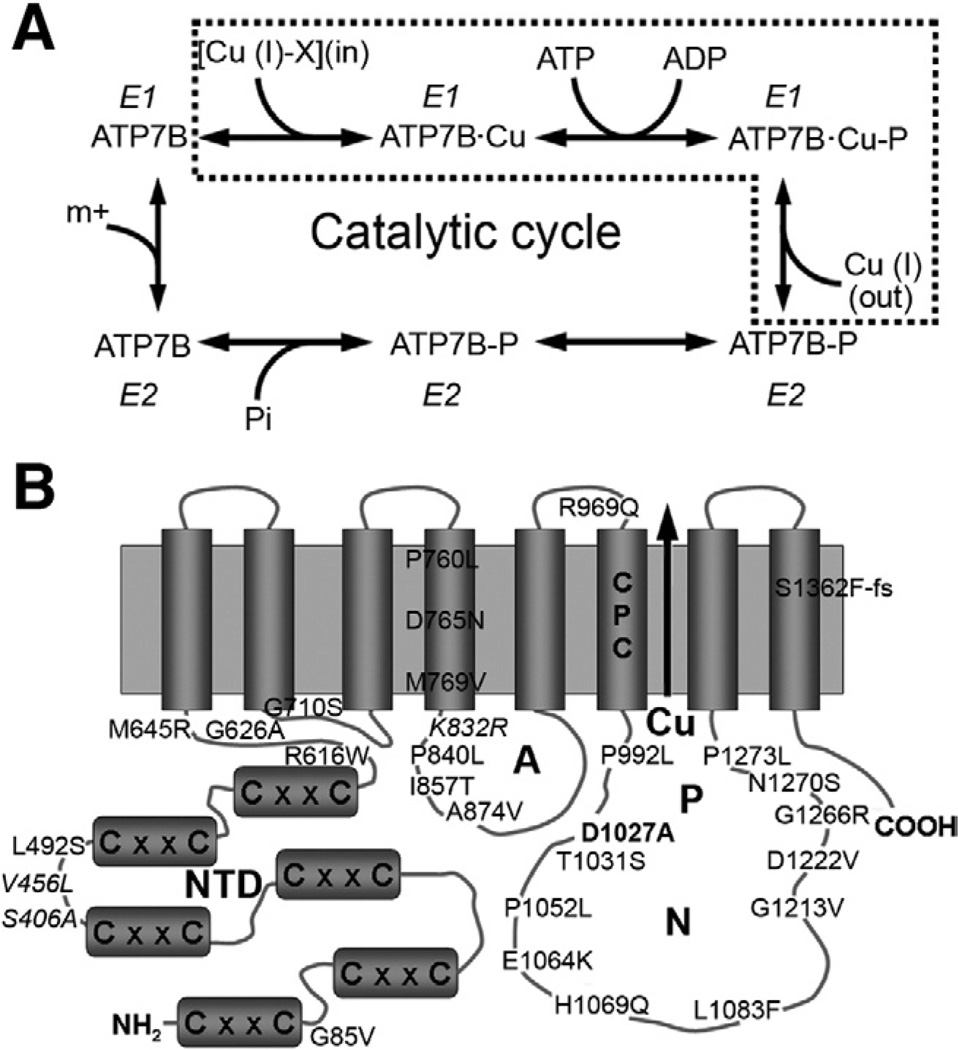
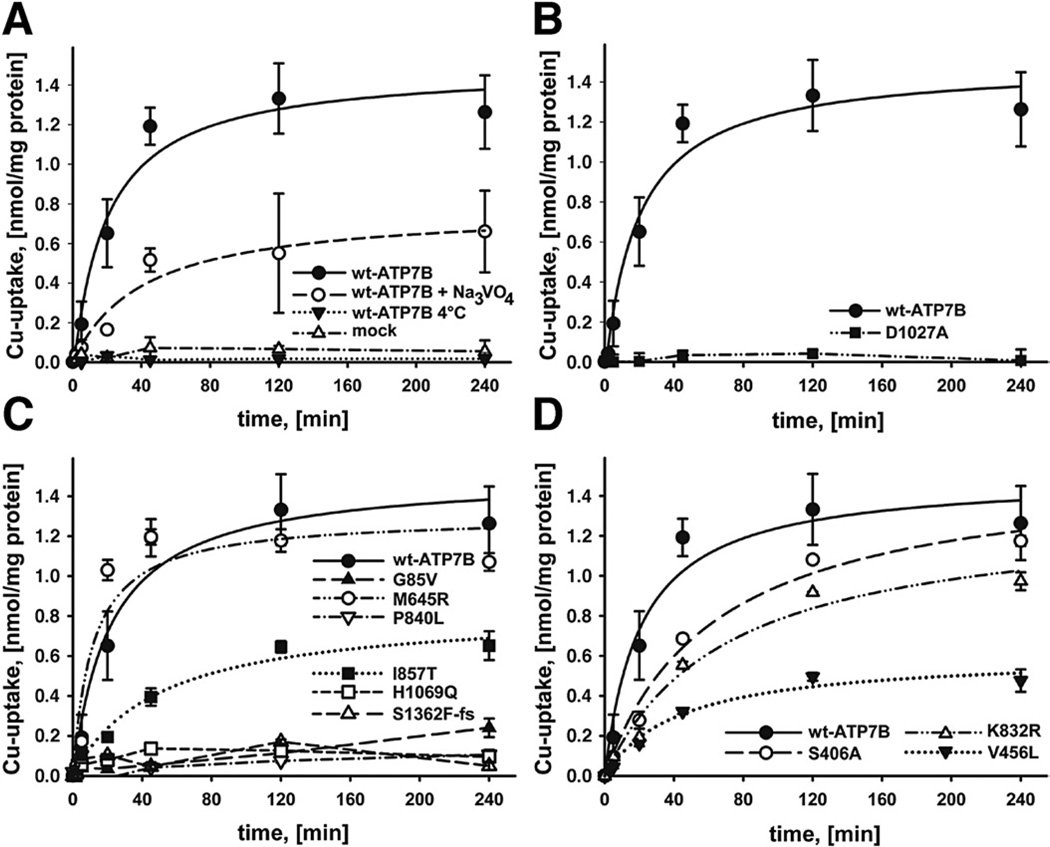
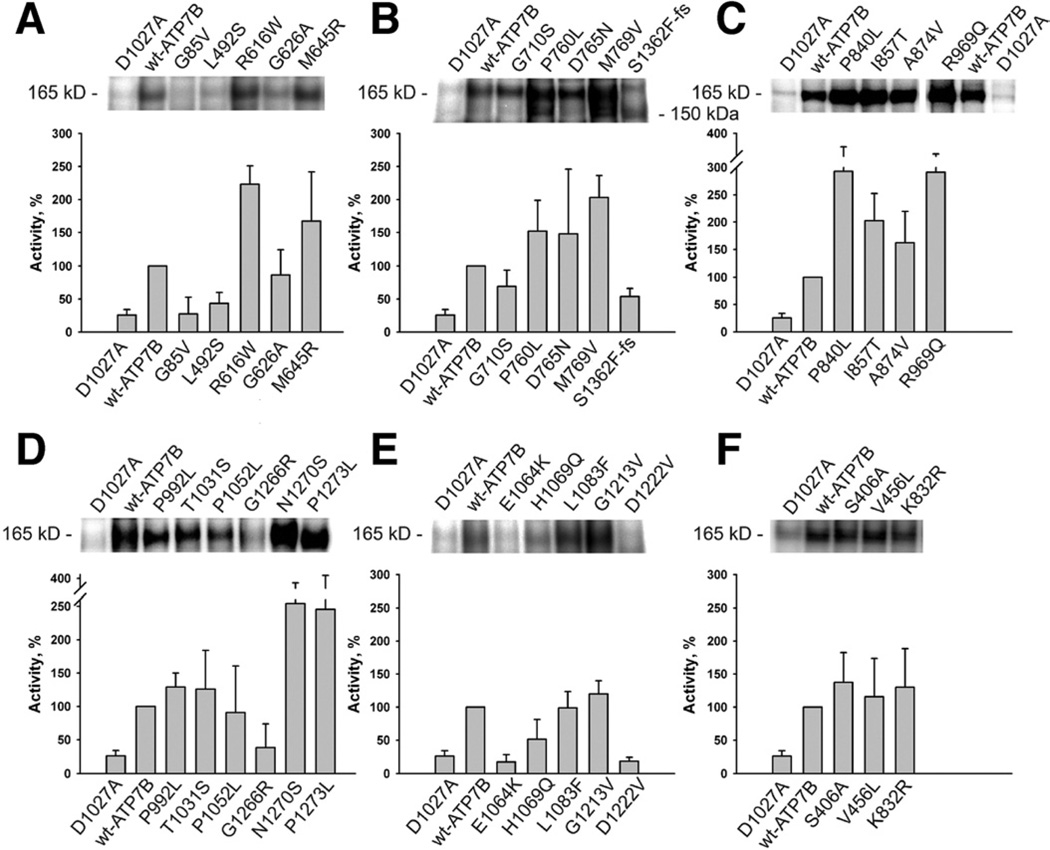
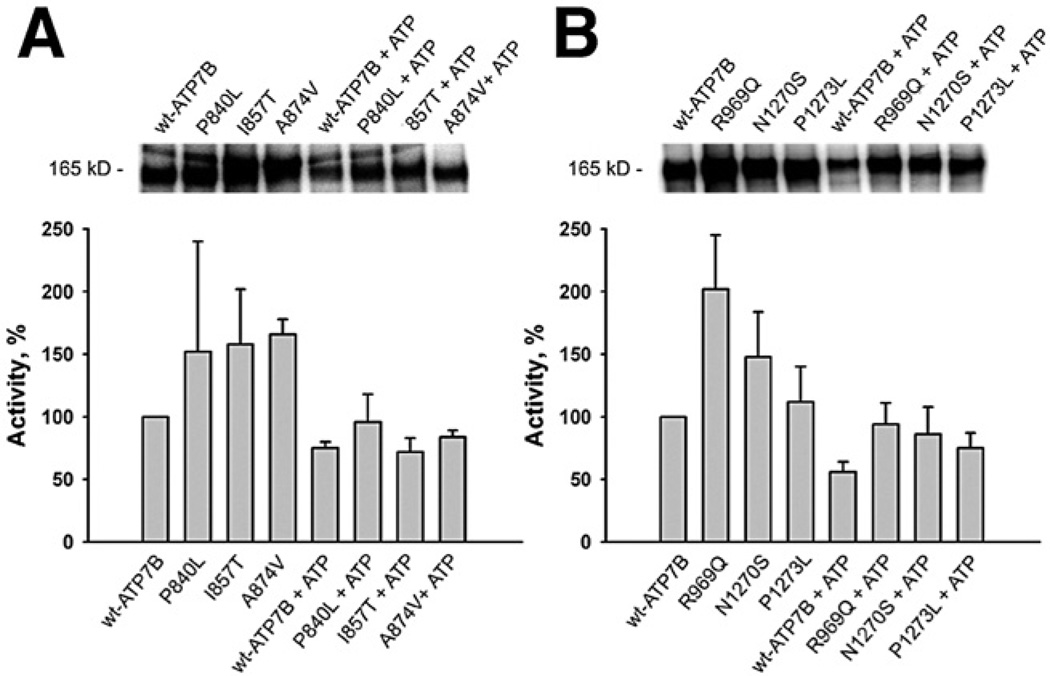
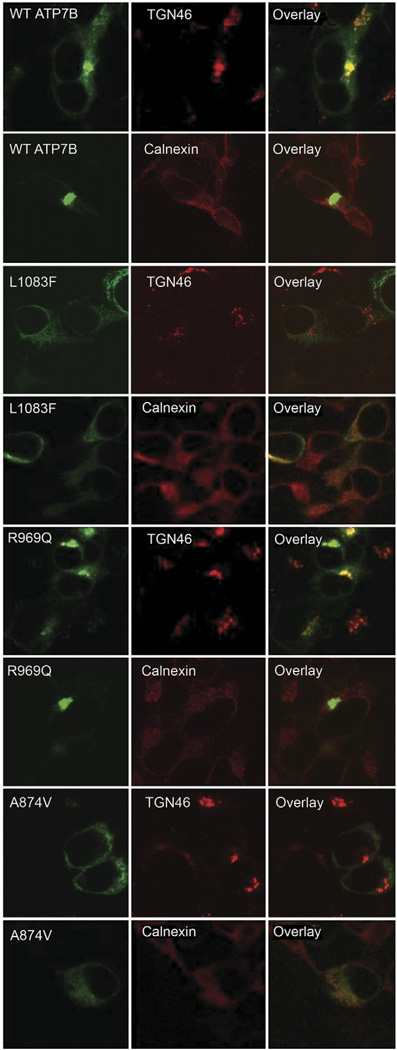
Methods: We analyzed 28 variants of ATP7B from patients with Wilson disease that affected different functional domains; the gene products were expressed using the baculovirus expression system in Sf9 cells. Protein function was analyzed by measuring catalytic activity and copper ((64)Cu) transport into vesicles. We studied intracellular localization of variants of ATP7B that had measurable transport activities and were tagged with green fluorescent protein in mammalian cells using confocal laser scanning microscopy.
Results: Properties of ATP7B variants with pathogenic amino-acid substitution varied greatly even if substitutions were in the same functional domain. Some variants had complete loss of catalytic and transport activity, whereas others lost transport activity but retained phosphor-intermediate formation or had partial losses of activity. In mammalian cells, transport-competent variants differed in stability and subcellular localization.
Conclusions: Variants in ATP7B associated with Wilson disease disrupt the protein's transport activity, result in its mislocalization, and reduce its stability. Single assays are insufficient to accurately predict the effects of ATP7B variants the function of its product and development of Wilson disease. These findings will contribute to our understanding of genotype-phenotype correlation and mechanisms of disease pathogenesis.
Copyright © 2012 AGA Institute. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflicts of interest
The authors disclose no conflicts.
Figures
Comment in
-
Defining Wilson disease phenotypes: from the patient to the bench and back again.Gastroenterology. 2012 Apr;142(4):692-6. doi: 10.1053/j.gastro.2012.02.035. Gastroenterology. 2012. PMID: 22480881 No abstract available.
References
-
- Pfeiffer RF. Wilson’s disease. Semin Neurol. 2007;27:123–132. - PubMed
-
- Huster D. Wilson disease. Best Pract Res Clin Gastroenterol. 2010;24:531–539. - PubMed
-
- Riordan SM, Williams R. The Wilson’s disease gene and phenotypic diversity. J Hepatol. 2001;34:165–171. - PubMed
-
- Lorincz MT. Neurologic Wilson’s disease. Ann N Y Acad Sci. 2010;1184:173–187. - PubMed
-
- Kegley KM, Sellers MA, Ferber MJ, et al. Fulminant Wilson’s disease requiring liver transplantation in one monozygotic twin despite identical genetic mutation. Am J Transplant. 2010;10:1325–1329. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
