

Mitochondrial permeability transition in rat hepatocytes after anoxia/reoxygenation: role of Ca2+-dependent mitochondrial formation of reactive oxygen species
- PMID: 22241863
- PMCID: PMC3330780
- DOI: 10.1152/ajpgi.00082.2011
Mitochondrial permeability transition in rat hepatocytes after anoxia/reoxygenation: role of Ca2+-dependent mitochondrial formation of reactive oxygen species
Abstract
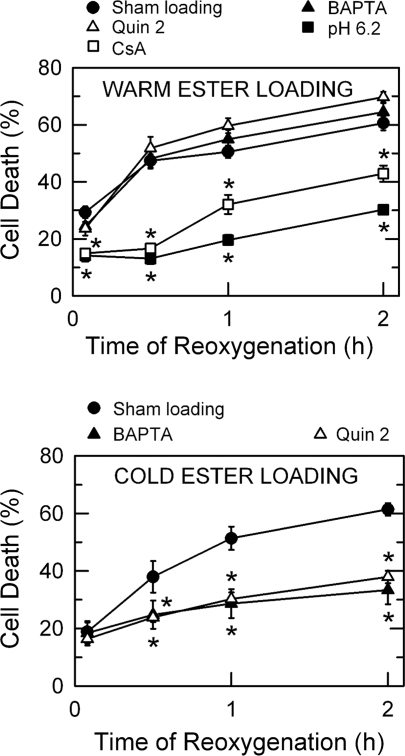
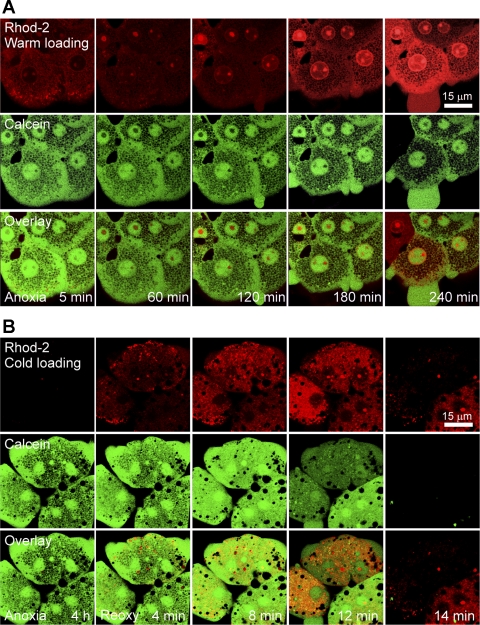
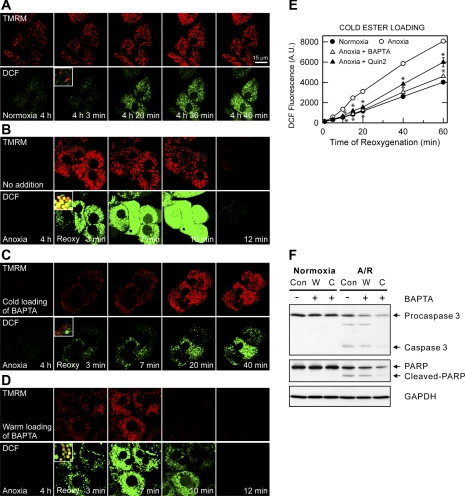
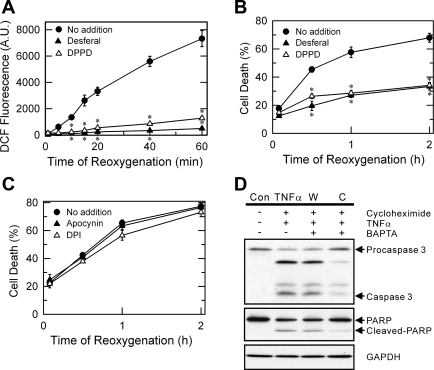
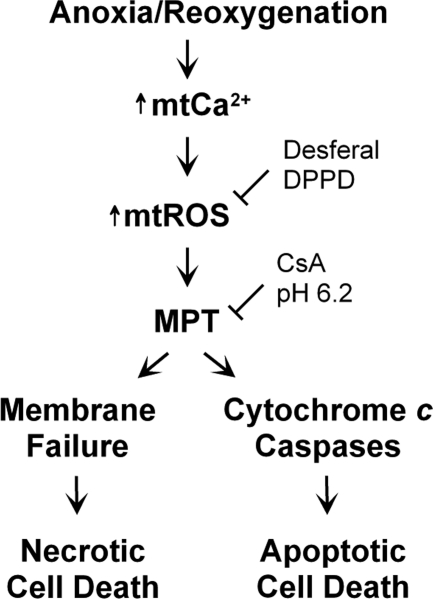
Onset of the mitochondrial permeability transition (MPT) is the penultimate event leading to lethal cellular ischemia-reperfusion injury, but the mechanisms precipitating the MPT after reperfusion remain unclear. Here, we investigated the role of mitochondrial free Ca(2+) and reactive oxygen species (ROS) in pH- and MPT-dependent reperfusion injury to hepatocytes. Cultured rat hepatocytes were incubated in anoxic Krebs-Ringer-HEPES buffer at pH 6.2 for 4 h and then reoxygenated at pH 7.4 to simulate ischemia-reperfusion. Some cells were loaded with the Ca(2+) chelators, BAPTA/AM and 2-[(2-bis-[carboxymethyl]aono-5-methoxyphenyl)-methyl-6-methoxy-8-bis[carboxymethyl]aminoquinoline, either by a cold loading protocol for intramitochondrial loading or by warm incubation for cytosolic loading. Cell death was assessed by propidium iodide fluorometry and immunoblotting. Mitochondrial Ca(2+), inner membrane permeability, membrane potential, and ROS formation were monitored with Rhod-2, calcein, tetramethylrhodamine methylester, and dihydrodichlorofluorescein, respectively. Necrotic cell death increased after reoxygenation. Necrosis was blocked by 1 μM cyclosporin A, an MPT inhibitor, and by reoxygenation at pH 6.2. Confocal imaging of Rhod-2, calcein, and dichlorofluorescein revealed that an increase of mitochondrial Ca(2+) and ROS preceded onset of the MPT after reoxygenation. Intramitochondrial Ca(2+) chelation, but not cytosolic Ca(2+) chelation, prevented ROS formation and subsequent necrotic and apoptotic cell death. Reoxygenation with the antioxidants, desferal or diphenylphenylenediamine, also suppressed MPT-mediated cell death. However, inhibition of cytosolic ROS by apocynin or diphenyleneiodonium chloride failed to prevent reoxygenation-induced cell death. In conclusion, Ca(2+)-dependent mitochondrial ROS formation is the molecular signal culminating in onset of the MPT after reoxygenation of anoxic hepatocytes, leading to cell death.
Figures
References
-
- Battaglia V, Grancara S, Satriano J, Saccoccio S, Agostinelli E, Toninello A. Agmatine prevents the Ca2+-dependent induction of permeability transition in rat brain mitochondria. Amino Acids 38: 431–437, 2010 - PubMed
-
- Berger NA, Petzold SJ. Identification of minimal size requirements of DNA for activation of poly(ADP-ribose) polymerase. Biochemistry 24: 4352–4355, 1985 - PubMed
-
- Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev 79: 1127–1155, 1999 - PubMed
-
- Bhogal RH, Curbishley SM, Weston CJ, Adams DH, Afford SC. Reactive oxygen species mediate human hepatocyte injury during hypoxia/reoxygenation. Liver Transpl 16: 1303–1313, 2010 - PubMed
-
- Black D, Bird MA, Samson CM, Lyman S, Lange PA, Schrum LW, Qian T, Lemasters JJ, Brenner DA, Rippe RA, Behrns KE. Primary cirrhotic hepatocytes resist TGFbeta-induced apoptosis through a ROS-dependent mechanism. J Hepatol 40: 942–951, 2004 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous