

Protecting High Energy Barriers: A New Equation to Regulate Boost Energy in Accelerated Molecular Dynamics Simulations
- PMID: 22241967
- PMCID: PMC3254191
- DOI: 10.1021/ct200615k
Protecting High Energy Barriers: A New Equation to Regulate Boost Energy in Accelerated Molecular Dynamics Simulations
Abstract
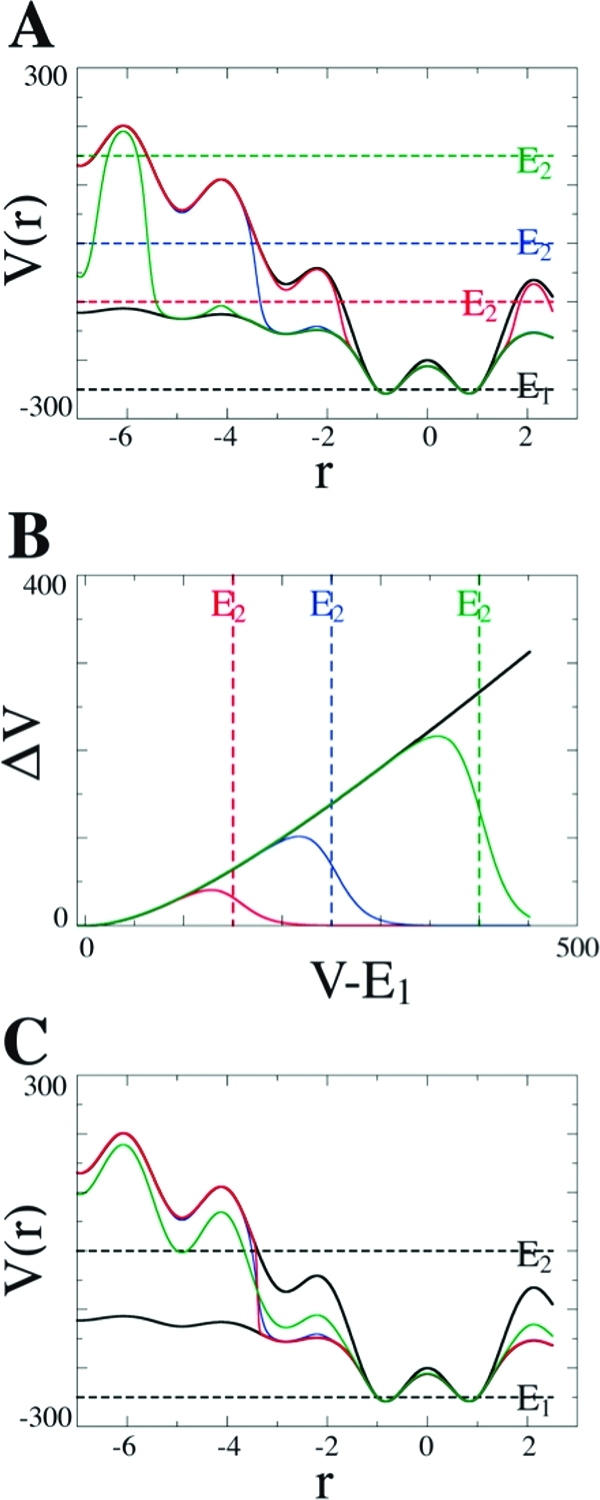
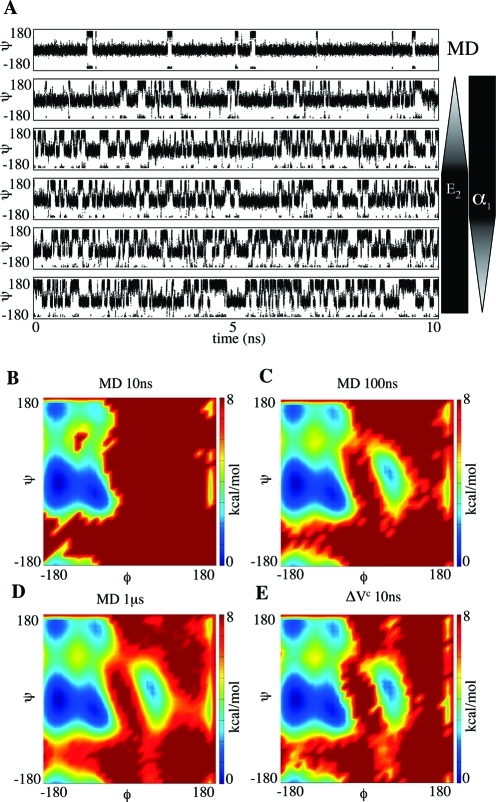
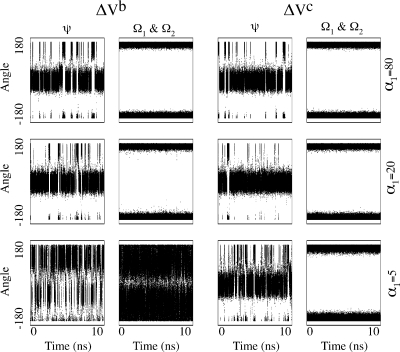
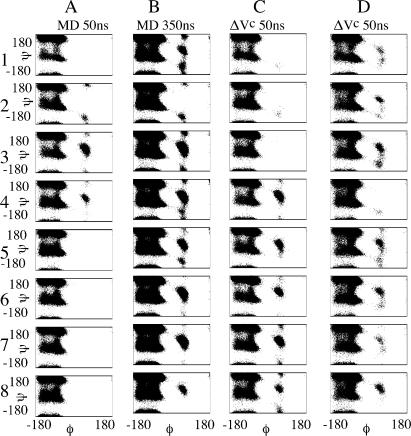
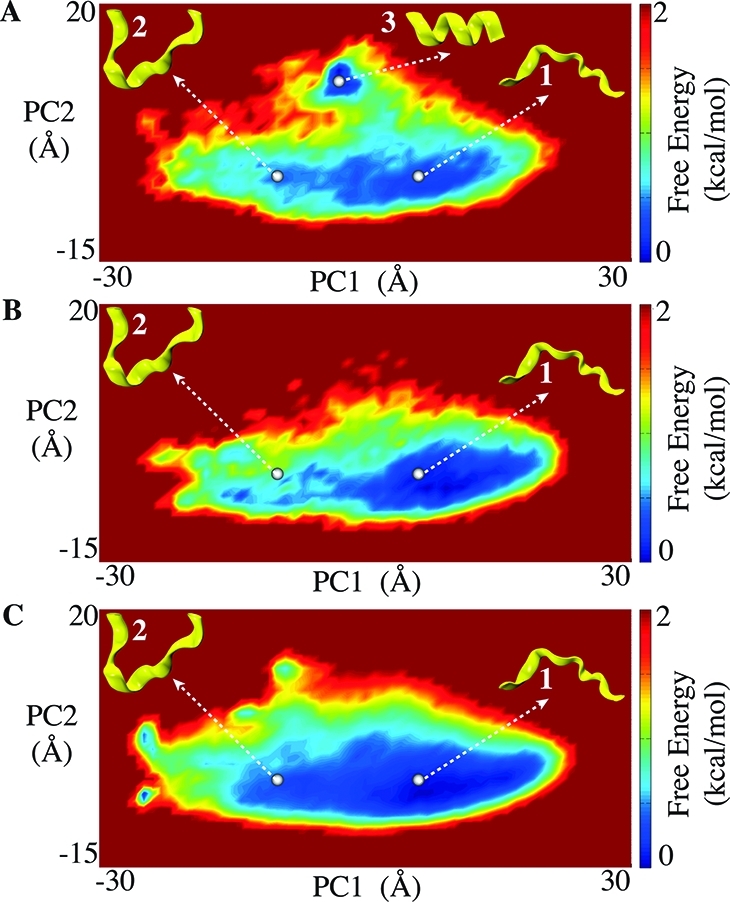
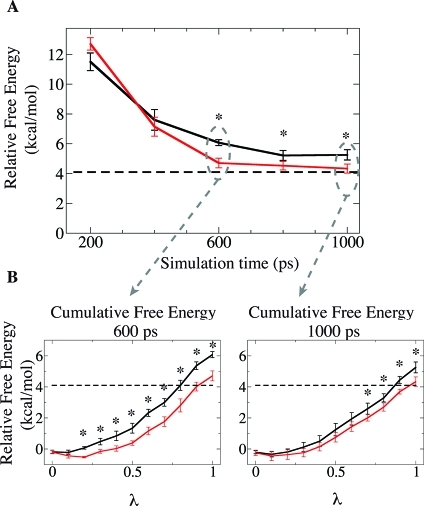
Molecular dynamics (MD) is one of the most common tools in computational chemistry. Recently, our group has employed accelerated molecular dynamics (aMD) to improve the conformational sampling over conventional molecular dynamics techniques. In the original aMD implementation, sampling is greatly improved by raising energy wells below a predefined energy level. Recently, our group presented an alternative aMD implementation where simulations are accelerated by lowering energy barriers of the potential energy surface. When coupled with thermodynamic integration simulations, this implementation showed very promising results. However, when applied to large systems, such as proteins, the simulation tends to be biased to high energy regions of the potential landscape. The reason for this behavior lies in the boost equation used since the highest energy barriers are dramatically more affected than the lower ones. To address this issue, in this work, we present a new boost equation that prevents oversampling of unfavorable high energy conformational states. The new boost potential provides not only better recovery of statistics throughout the simulation but also enhanced sampling of statistically relevant regions in explicit solvent MD simulations.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources