

A mathematical methodology for determining the temporal order of pathway alterations arising during gliomagenesis
- PMID: 22241976
- PMCID: PMC3252265
- DOI: 10.1371/journal.pcbi.1002337
A mathematical methodology for determining the temporal order of pathway alterations arising during gliomagenesis
Abstract
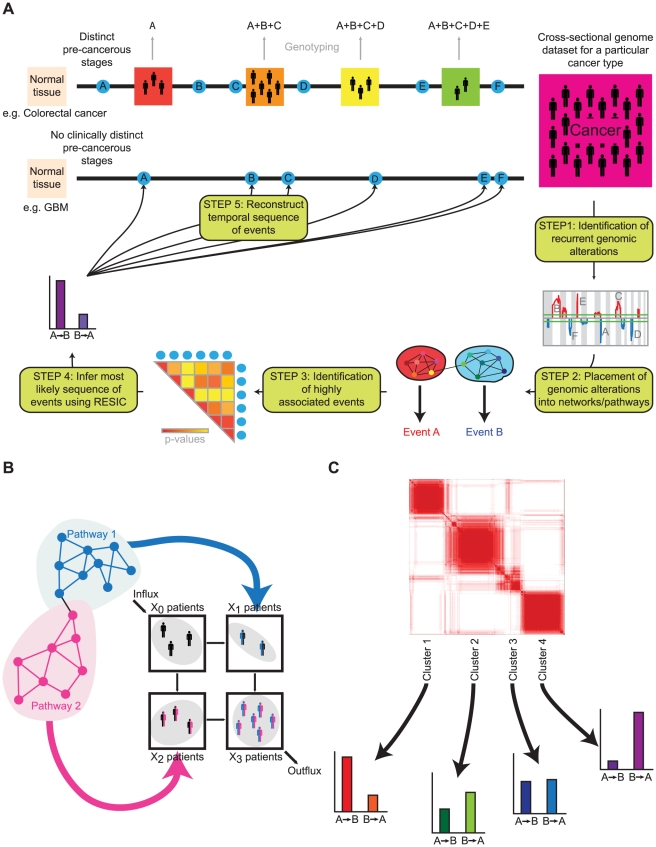
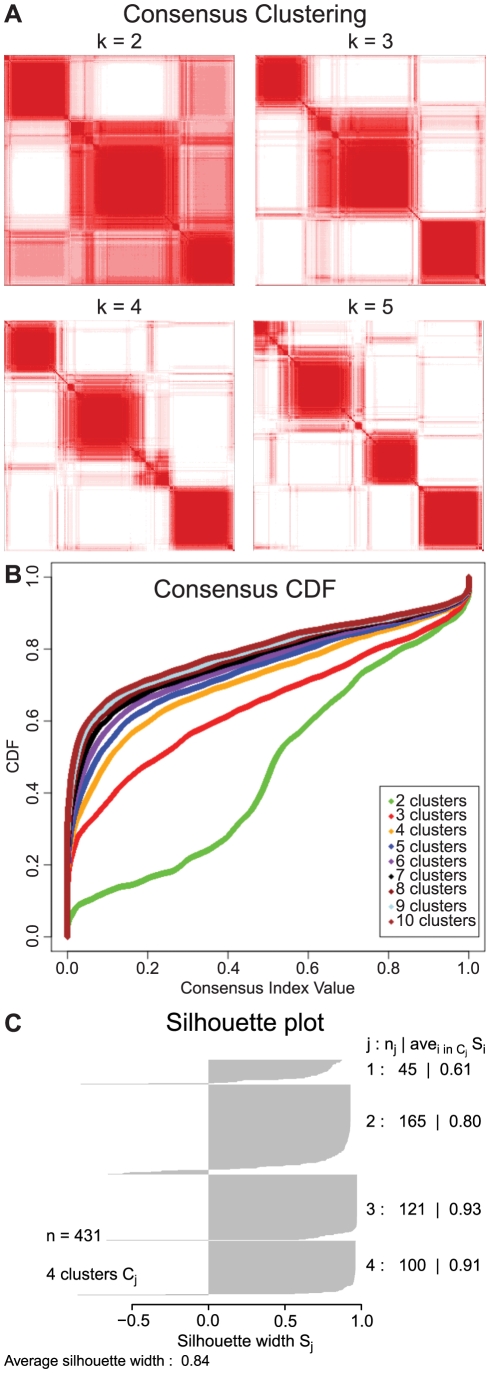
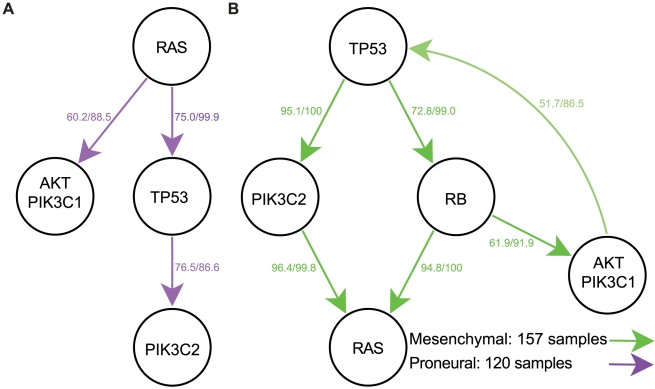
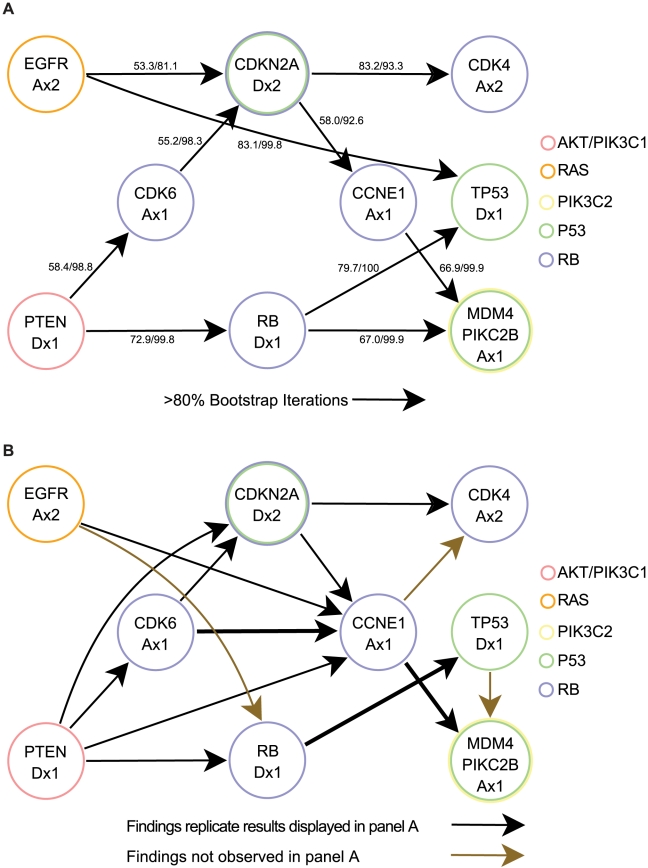
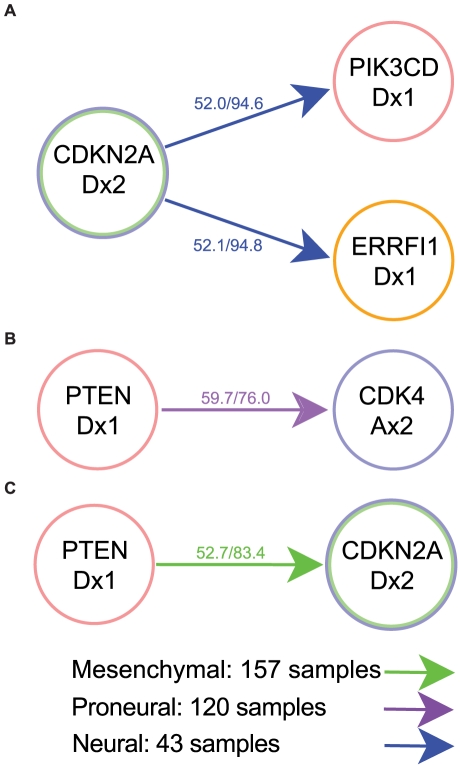
Human cancer is caused by the accumulation of genetic alterations in cells. Of special importance are changes that occur early during malignant transformation because they may result in oncogene addiction and thus represent promising targets for therapeutic intervention. We have previously described a computational approach, called Retracing the Evolutionary Steps in Cancer (RESIC), to determine the temporal sequence of genetic alterations during tumorigenesis from cross-sectional genomic data of tumors at their fully transformed stage. Since alterations within a set of genes belonging to a particular signaling pathway may have similar or equivalent effects, we applied a pathway-based systems biology approach to the RESIC methodology. This method was used to determine whether alterations of specific pathways develop early or late during malignant transformation. When applied to primary glioblastoma (GBM) copy number data from The Cancer Genome Atlas (TCGA) project, RESIC identified a temporal order of pathway alterations consistent with the order of events in secondary GBMs. We then further subdivided the samples into the four main GBM subtypes and determined the relative contributions of each subtype to the overall results: we found that the overall ordering applied for the proneural subtype but differed for mesenchymal samples. The temporal sequence of events could not be identified for neural and classical subtypes, possibly due to a limited number of samples. Moreover, for samples of the proneural subtype, we detected two distinct temporal sequences of events: (i) RAS pathway activation was followed by TP53 inactivation and finally PI3K2 activation, and (ii) RAS activation preceded only AKT activation. This extension of the RESIC methodology provides an evolutionary mathematical approach to identify the temporal sequence of pathway changes driving tumorigenesis and may be useful in guiding the understanding of signaling rearrangements in cancer development.
© 2012 Cheng et al.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

Similar articles
-
A mathematical framework to determine the temporal sequence of somatic genetic events in cancer.Proc Natl Acad Sci U S A. 2010 Oct 12;107(41):17604-9. doi: 10.1073/pnas.1009117107. Epub 2010 Sep 23. Proc Natl Acad Sci U S A. 2010. PMID: 20864632 Free PMC article.
-
miR-34a repression in proneural malignant gliomas upregulates expression of its target PDGFRA and promotes tumorigenesis.PLoS One. 2012;7(3):e33844. doi: 10.1371/journal.pone.0033844. Epub 2012 Mar 27. PLoS One. 2012. PMID: 22479456 Free PMC article.
-
Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations.PLoS One. 2009 Nov 13;4(11):e7752. doi: 10.1371/journal.pone.0007752. PLoS One. 2009. PMID: 19915670 Free PMC article.
-
Molecular and Genomic Alterations in Glioblastoma Multiforme.Am J Pathol. 2015 Jul;185(7):1820-33. doi: 10.1016/j.ajpath.2015.02.023. Epub 2015 May 11. Am J Pathol. 2015. PMID: 25976245 Review.
-
Targeting the AKT pathway in glioblastoma.Curr Pharm Des. 2011;17(23):2411-20. doi: 10.2174/138161211797249224. Curr Pharm Des. 2011. PMID: 21827416 Review.
Cited by
-
Computational medicine: translating models to clinical care.Sci Transl Med. 2012 Oct 31;4(158):158rv11. doi: 10.1126/scitranslmed.3003528. Sci Transl Med. 2012. PMID: 23115356 Free PMC article.
-
Temporal ordering of cancer microarray data through a reinforcement learning based approach.PLoS One. 2013;8(4):e60883. doi: 10.1371/journal.pone.0060883. Epub 2013 Apr 2. PLoS One. 2013. PMID: 23565283 Free PMC article.
-
Motion, fixation probability and the choice of an evolutionary process.PLoS Comput Biol. 2019 Aug 5;15(8):e1007238. doi: 10.1371/journal.pcbi.1007238. eCollection 2019 Aug. PLoS Comput Biol. 2019. PMID: 31381556 Free PMC article.
-
Phylogenetic analysis of multiprobe fluorescence in situ hybridization data from tumor cell populations.Bioinformatics. 2013 Jul 1;29(13):i189-98. doi: 10.1093/bioinformatics/btt205. Bioinformatics. 2013. PMID: 23812984 Free PMC article.
-
Identification of constrained cancer driver genes based on mutation timing.PLoS Comput Biol. 2015 Jan 8;11(1):e1004027. doi: 10.1371/journal.pcbi.1004027. eCollection 2015 Jan. PLoS Comput Biol. 2015. PMID: 25569148 Free PMC article.
References
-
- Sjöblom T, Jones S, Wood LD, Parsons DW, Lin J, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
