

Common variants show predicted polygenic effects on height in the tails of the distribution, except in extremely short individuals
- PMID: 22242009
- PMCID: PMC3248463
- DOI: 10.1371/journal.pgen.1002439
Common variants show predicted polygenic effects on height in the tails of the distribution, except in extremely short individuals
Abstract
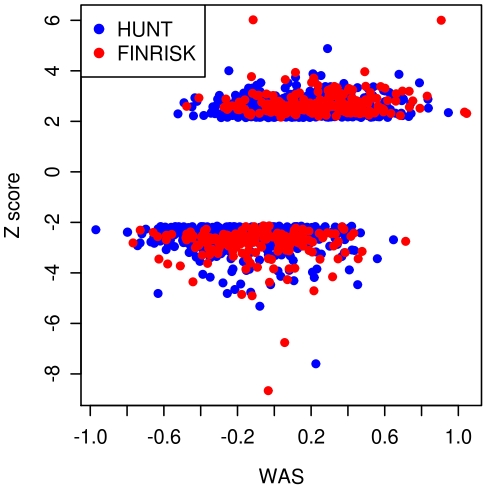
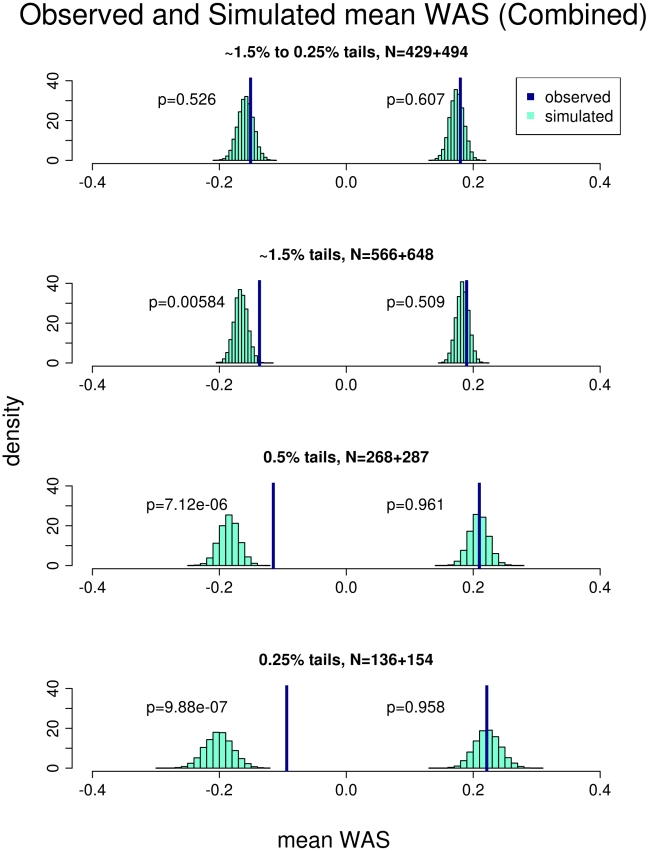
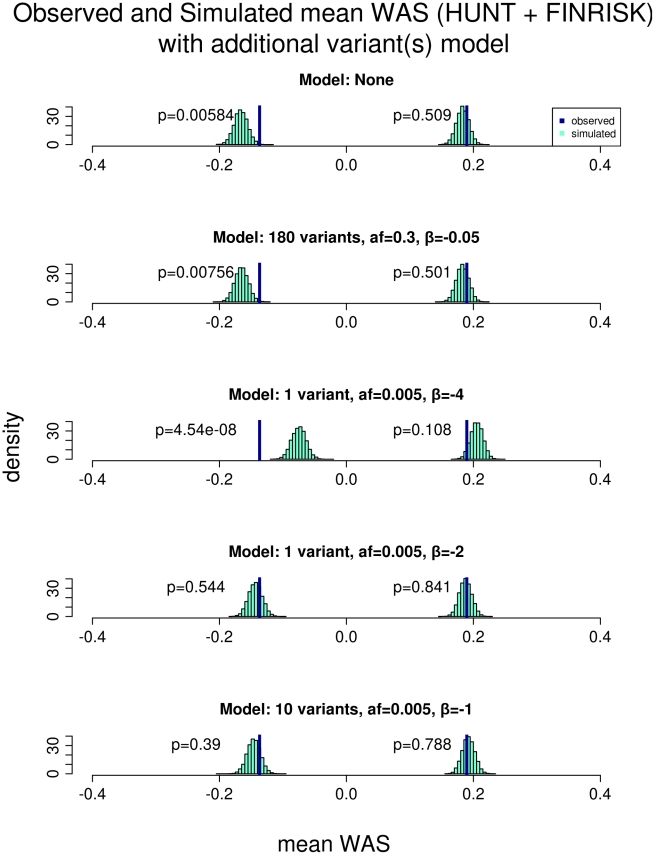
Common genetic variants have been shown to explain a fraction of the inherited variation for many common diseases and quantitative traits, including height, a classic polygenic trait. The extent to which common variation determines the phenotype of highly heritable traits such as height is uncertain, as is the extent to which common variation is relevant to individuals with more extreme phenotypes. To address these questions, we studied 1,214 individuals from the top and bottom extremes of the height distribution (tallest and shortest ∼1.5%), drawn from ∼78,000 individuals from the HUNT and FINRISK cohorts. We found that common variants still influence height at the extremes of the distribution: common variants (49/141) were nominally associated with height in the expected direction more often than is expected by chance (p<5×10⁻²⁸), and the odds ratios in the extreme samples were consistent with the effects estimated previously in population-based data. To examine more closely whether the common variants have the expected effects, we calculated a weighted allele score (WAS), which is a weighted prediction of height for each individual based on the previously estimated effect sizes of the common variants in the overall population. The average WAS is consistent with expectation in the tall individuals, but was not as extreme as expected in the shortest individuals (p<0.006), indicating that some of the short stature is explained by factors other than common genetic variation. The discrepancy was more pronounced (p<10⁻⁶) in the most extreme individuals (height<0.25 percentile). The results at the extreme short tails are consistent with a large number of models incorporating either rare genetic non-additive or rare non-genetic factors that decrease height. We conclude that common genetic variants are associated with height at the extremes as well as across the population, but that additional factors become more prominent at the shorter extreme.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet. 2010;11:415–425. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
