

Microenvironmental regulation by fibrillin-1
- PMID: 22242013
- PMCID: PMC3252277
- DOI: 10.1371/journal.pgen.1002425
Microenvironmental regulation by fibrillin-1
Abstract
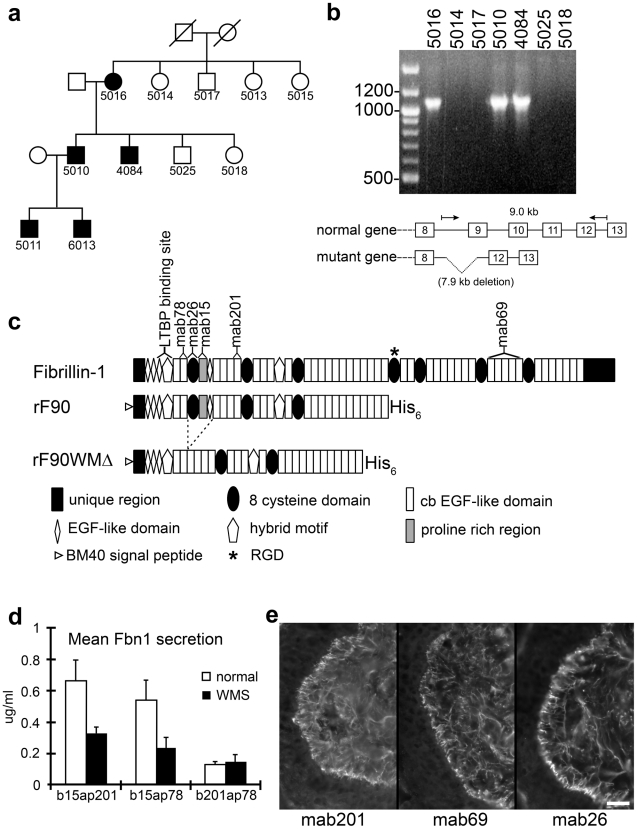
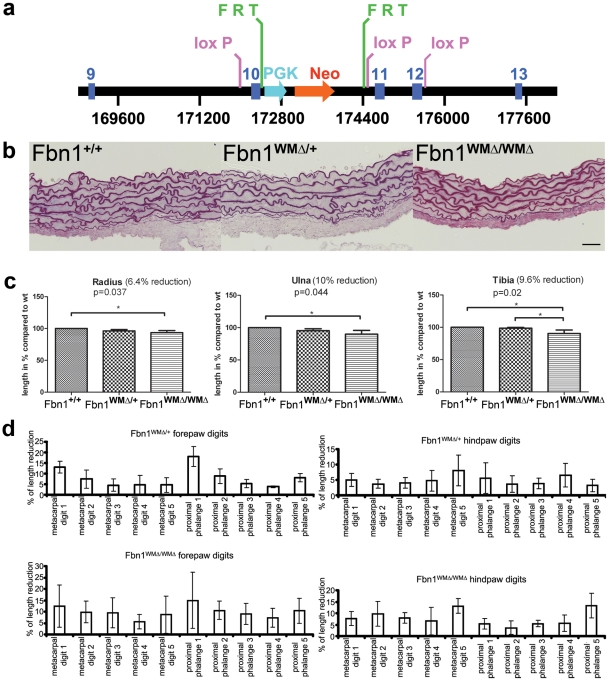
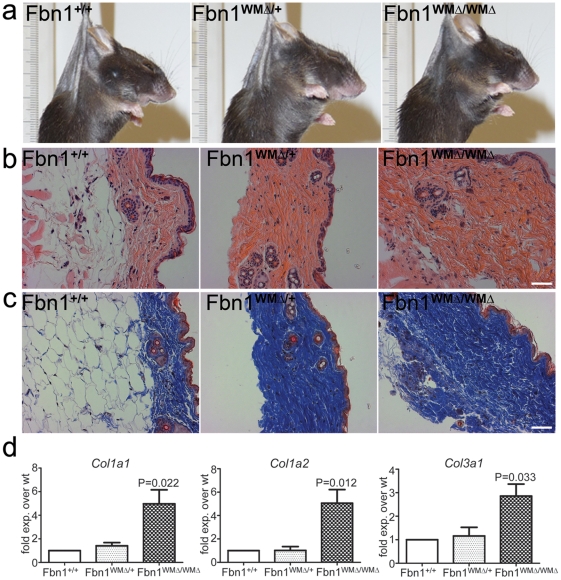
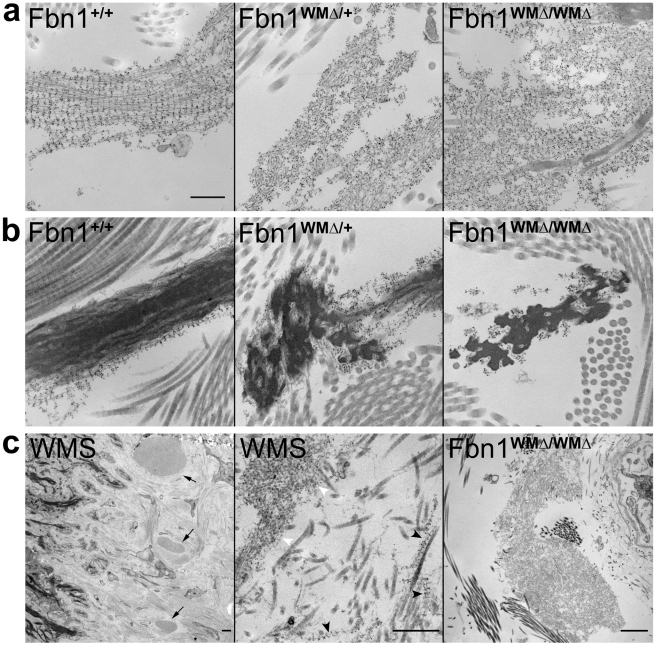
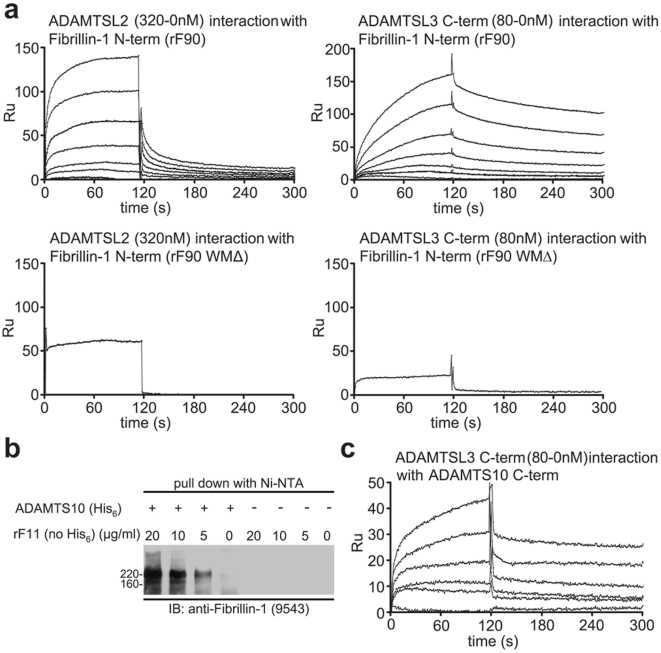
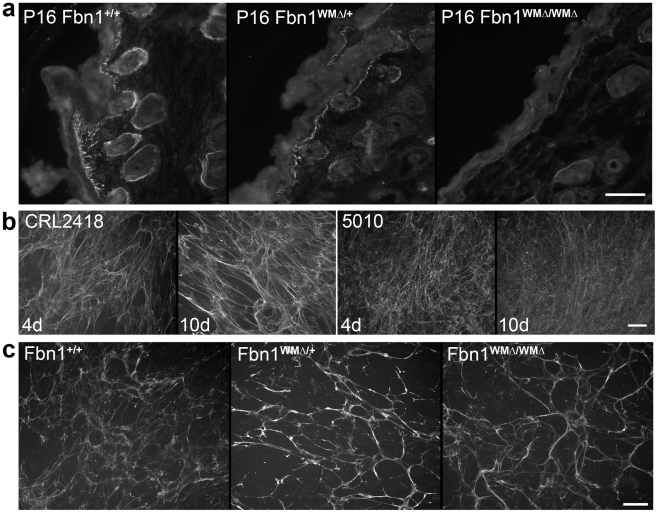
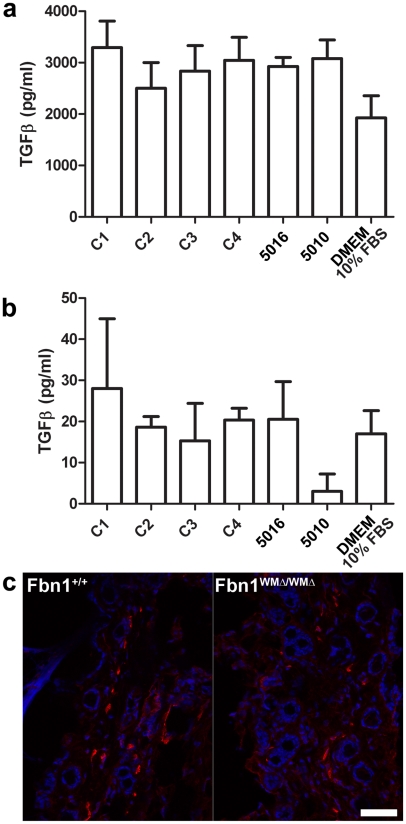
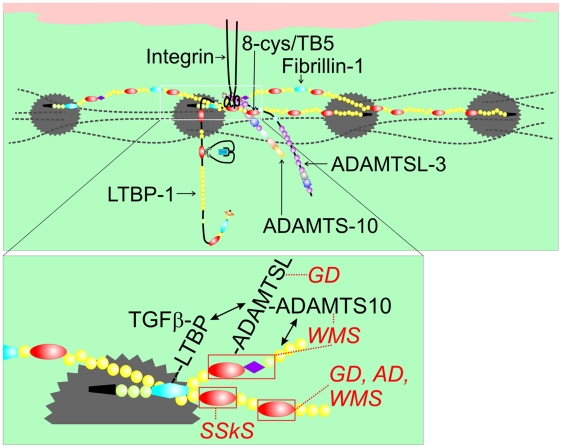
Fibrillin-1 is a ubiquitous extracellular matrix molecule that sequesters latent growth factor complexes. A role for fibrillin-1 in specifying tissue microenvironments has not been elucidated, even though the concept that fibrillin-1 provides extracellular control of growth factor signaling is currently appreciated. Mutations in FBN1 are mainly responsible for the Marfan syndrome (MFS), recognized by its pleiotropic clinical features including tall stature and arachnodactyly, aortic dilatation and dissection, and ectopia lentis. Each of the many different mutations in FBN1 known to cause MFS must lead to similar clinical features through common mechanisms, proceeding principally through the activation of TGFβ signaling. Here we show that a novel FBN1 mutation in a family with Weill-Marchesani syndrome (WMS) causes thick skin, short stature, and brachydactyly when replicated in mice. WMS mice confirm that this mutation does not cause MFS. The mutation deletes three domains in fibrillin-1, abolishing a binding site utilized by ADAMTSLIKE-2, -3, -6, and papilin. Our results place these ADAMTSLIKE proteins in a molecular pathway involving fibrillin-1 and ADAMTS-10. Investigations of microfibril ultrastructure in WMS humans and mice demonstrate that modulation of the fibrillin microfibril scaffold can influence local tissue microenvironments and link fibrillin-1 function to skin homeostasis and the regulation of dermal collagen production. Hence, pathogenetic mechanisms caused by dysregulated WMS microenvironments diverge from Marfan pathogenetic mechanisms, which lead to broad activation of TGFβ signaling in multiple tissues. We conclude that local tissue-specific microenvironments, affected in WMS, are maintained by a fibrillin-1 microfibril scaffold, modulated by ADAMTSLIKE proteins in concert with ADAMTS enzymes.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Missense mutations in FBN1 exons 41 and 42 cause Weill-Marchesani syndrome with thoracic aortic disease and Marfan syndrome.Am J Med Genet A. 2013 Sep;161A(9):2305-10. doi: 10.1002/ajmg.a.36044. Epub 2013 Jul 29. Am J Med Genet A. 2013. PMID: 23897642 Free PMC article.
-
Adamtsl2 deletion results in bronchial fibrillin microfibril accumulation and bronchial epithelial dysplasia--a novel mouse model providing insights into geleophysic dysplasia.Dis Model Mech. 2015 May;8(5):487-99. doi: 10.1242/dmm.017046. Epub 2015 Mar 11. Dis Model Mech. 2015. PMID: 25762570 Free PMC article.
-
Genetic and functional linkage between ADAMTS superfamily proteins and fibrillin-1: a novel mechanism influencing microfibril assembly and function.Cell Mol Life Sci. 2011 Oct;68(19):3137-48. doi: 10.1007/s00018-011-0780-9. Epub 2011 Aug 20. Cell Mol Life Sci. 2011. PMID: 21858451 Free PMC article. Review.
-
The fibrillin microfibril scaffold: A niche for growth factors and mechanosensation?Matrix Biol. 2015 Sep;47:3-12. doi: 10.1016/j.matbio.2015.05.002. Epub 2015 May 7. Matrix Biol. 2015. PMID: 25957947 Review.
-
Microfibrils: a cornerstone of extracellular matrix and a key to understand Marfan syndrome.Ital J Anat Embryol. 2009 Oct-Dec;114(4):201-24. Ital J Anat Embryol. 2009. PMID: 20578676 Review.
Cited by
-
Genetic models of fibrillinopathies.Genetics. 2024 Jan 3;226(1):iyad189. doi: 10.1093/genetics/iyad189. Genetics. 2024. PMID: 37972149 Free PMC article. Review.
-
Fibrillin microfibrils and elastic fibre proteins: Functional interactions and extracellular regulation of growth factors.Semin Cell Dev Biol. 2019 May;89:109-117. doi: 10.1016/j.semcdb.2018.07.016. Epub 2018 Jul 20. Semin Cell Dev Biol. 2019. PMID: 30016650 Free PMC article. Review.
-
Growth plate extracellular matrix defects and short stature in children.Ann Pediatr Endocrinol Metab. 2022 Dec;27(4):247-255. doi: 10.6065/apem.2244120.060. Epub 2022 Dec 31. Ann Pediatr Endocrinol Metab. 2022. PMID: 36567461 Free PMC article.
-
Cooperative Mechanism of ADAMTS/ ADAMTSL and Fibrillin-1 in the Marfan Syndrome and Acromelic Dysplasias.Front Genet. 2021 Nov 29;12:734718. doi: 10.3389/fgene.2021.734718. eCollection 2021. Front Genet. 2021. PMID: 34912367 Free PMC article. Review.
-
NMR spectroscopic and bioinformatic analyses of the LTBP1 C-terminus reveal a highly dynamic domain organisation.PLoS One. 2014 Jan 29;9(1):e87125. doi: 10.1371/journal.pone.0087125. eCollection 2014. PLoS One. 2014. PMID: 24489852 Free PMC article.
References
-
- McKusick VA. The Weill-Marchesani syndrome. In: McKusick VA, editor. Heritable disorders of connective tissue. 4th edition. St. Louis, MO: CV Mosby Company; 1972. pp. 282–291.
-
- Wirtz MK, Samples JR, Kramer PL, Rust K, Yount J, et al. Weill-Marchesani syndrome–possible linkage of the autosomal dominant form to 15q21.1. Am J Med Genet. 1996;65:68–75. - PubMed
-
- Faivre L, Megarbane A, Alswaid A, Zylberbeg L, Aldohayan N, et al. Homozygosity mapping of a Weill-Marchesani syndrome locus to chromosome 19p13.3-p13.2. Hum Genet. 2002;110:366–370. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
