

Genome-wide assessment of AU-rich elements by the AREScore algorithm
- PMID: 22242014
- PMCID: PMC3252268
- DOI: 10.1371/journal.pgen.1002433
Genome-wide assessment of AU-rich elements by the AREScore algorithm
Abstract
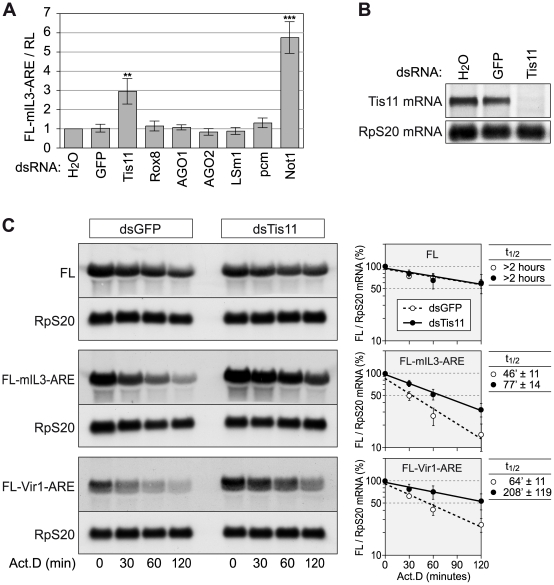
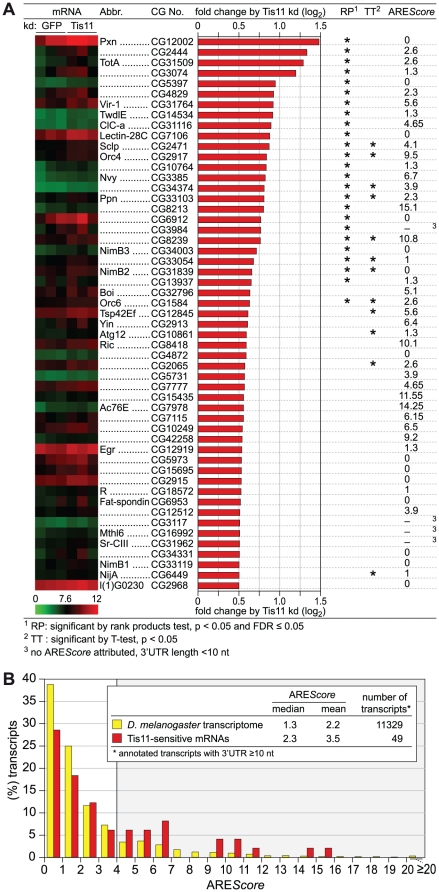
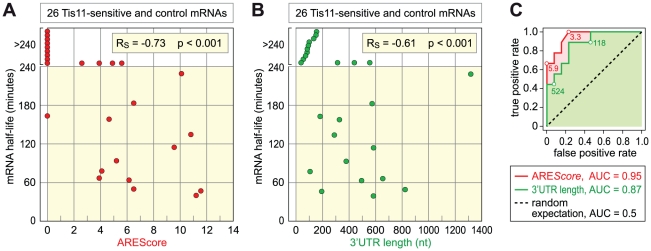
In mammalian cells, AU-rich elements (AREs) are well known regulatory sequences located in the 3' untranslated region (UTR) of many short-lived mRNAs. AREs cause mRNAs to be degraded rapidly and thereby suppress gene expression at the posttranscriptional level. Based on the number of AUUUA pentamers, their proximity, and surrounding AU-rich regions, we generated an algorithm termed AREScore that identifies AREs and provides a numerical assessment of their strength. By analyzing the AREScore distribution in the transcriptomes of 14 metazoan species, we provide evidence that AREs were selected for in several vertebrates and Drosophila melanogaster. We then measured mRNA expression levels genome-wide to address the importance of AREs in SL2 cells derived from D. melanogaster hemocytes. Tis11, a zinc finger RNA-binding protein homologous to mammalian tristetraprolin, was found to target ARE-containing reporter mRNAs for rapid degradation in SL2 cells. Drosophila mRNAs whose expression is elevated upon knock down of Tis11 were found to have higher AREScores. Moreover high AREScores correlate with reduced mRNA expression levels on a genome-wide scale. The precise measurement of degradation rates for 26 Drosophila mRNAs revealed that the AREScore is a very good predictor of short-lived mRNAs. Taken together, this study introduces AREScore as a simple tool to identify ARE-containing mRNAs and provides compelling evidence that AREs are widespread regulatory elements in Drosophila.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures




Similar articles
-
The Drosophila Tis11 protein and its effects on mRNA expression in flies.J Biol Chem. 2014 Dec 19;289(51):35042-60. doi: 10.1074/jbc.M114.593491. Epub 2014 Oct 23. J Biol Chem. 2014. PMID: 25342740 Free PMC article.
-
Drosophila eyes absent is a novel mRNA target of the tristetraprolin (TTP) protein DTIS11.Int J Biol Sci. 2012;8(5):606-19. doi: 10.7150/ijbs.3782. Epub 2012 Apr 20. Int J Biol Sci. 2012. PMID: 22553461 Free PMC article.
-
Rapid proteasomal degradation of posttranscriptional regulators of the TIS11/tristetraprolin family is induced by an intrinsically unstructured region independently of ubiquitination.Mol Cell Biol. 2014 Dec 1;34(23):4315-28. doi: 10.1128/MCB.00643-14. Epub 2014 Sep 22. Mol Cell Biol. 2014. PMID: 25246635 Free PMC article.
-
Multiple functions of tristetraprolin/TIS11 RNA-binding proteins in the regulation of mRNA biogenesis and degradation.Cell Mol Life Sci. 2013 Jun;70(12):2031-44. doi: 10.1007/s00018-012-1150-y. Epub 2012 Sep 12. Cell Mol Life Sci. 2013. PMID: 22968342 Free PMC article. Review.
-
Control of cytokine mRNA expression by RNA-binding proteins and microRNAs.J Dent Res. 2012 Jul;91(7):651-8. doi: 10.1177/0022034512437372. Epub 2012 Feb 1. J Dent Res. 2012. PMID: 22302144 Free PMC article. Review.
Cited by
-
dTIS11 Protein-dependent polysomal deadenylation is the key step in AU-rich element-mediated mRNA decay in Drosophila cells.J Biol Chem. 2012 Oct 12;287(42):35527-35538. doi: 10.1074/jbc.M112.356188. Epub 2012 Aug 29. J Biol Chem. 2012. PMID: 22932903 Free PMC article.
-
The Drosophila Tis11 protein and its effects on mRNA expression in flies.J Biol Chem. 2014 Dec 19;289(51):35042-60. doi: 10.1074/jbc.M114.593491. Epub 2014 Oct 23. J Biol Chem. 2014. PMID: 25342740 Free PMC article.
-
Genome-wide analysis of TIAR RNA ligands in mouse macrophages before and after LPS stimulation.Genom Data. 2016 Feb 8;7:297-300. doi: 10.1016/j.gdata.2016.02.007. eCollection 2016 Mar. Genom Data. 2016. PMID: 26981431 Free PMC article.
-
mRNA decay pre-complex assembly drives timely cell-state transitions during differentiation.Cell Rep. 2025 Jan 28;44(1):115138. doi: 10.1016/j.celrep.2024.115138. Epub 2024 Dec 30. Cell Rep. 2025. PMID: 39739530 Free PMC article.
-
TORn about SASP regulation.Cell Cycle. 2015;14(24):3771-2. doi: 10.1080/15384101.2015.1105694. Cell Cycle. 2015. PMID: 26697828 Free PMC article. No abstract available.
References
-
- Moore MJ. From birth to death: the complex lives of eukaryotic mRNAs. Science. 2005;309:1514–1518. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
