

Molecular genetic studies of complex phenotypes
- PMID: 22243791
- PMCID: PMC3259530
- DOI: 10.1016/j.trsl.2011.08.001
Molecular genetic studies of complex phenotypes
Abstract
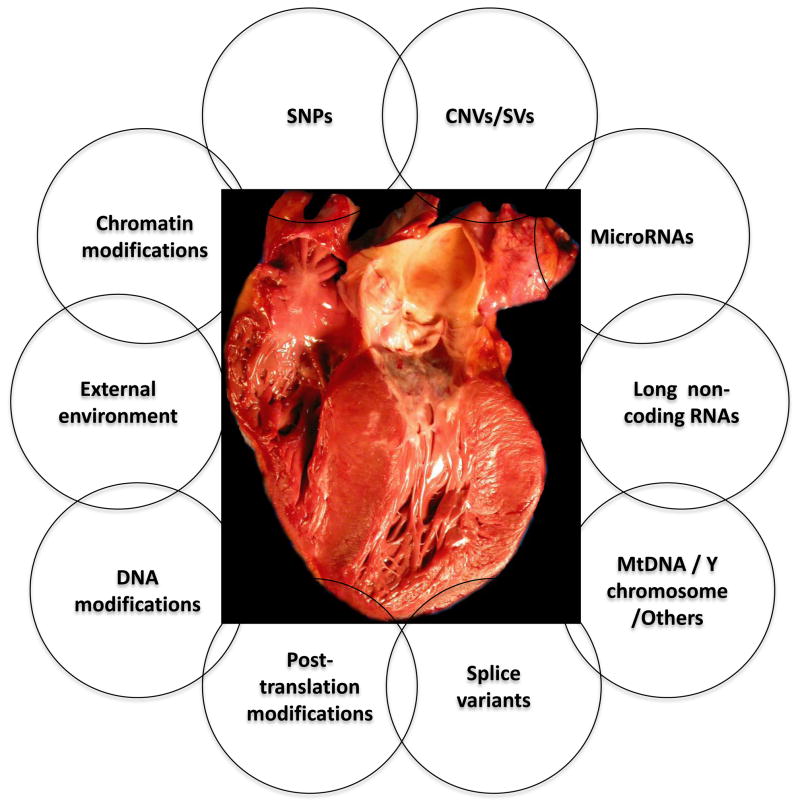
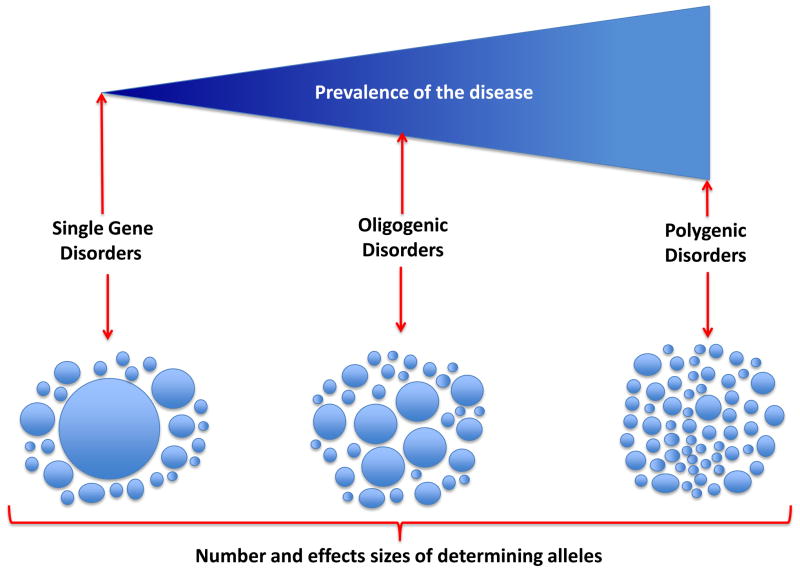
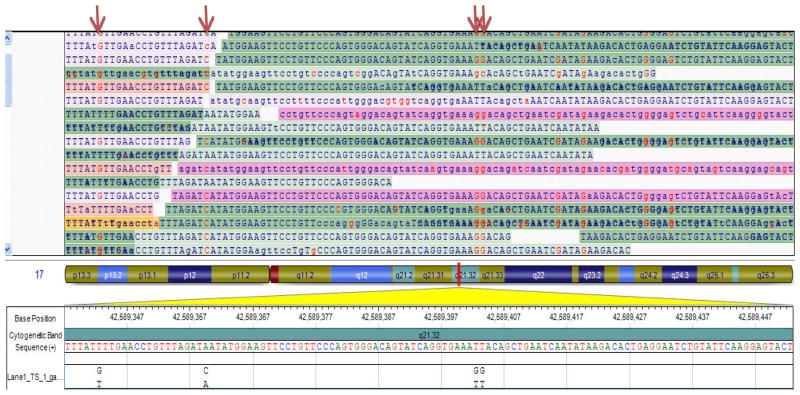
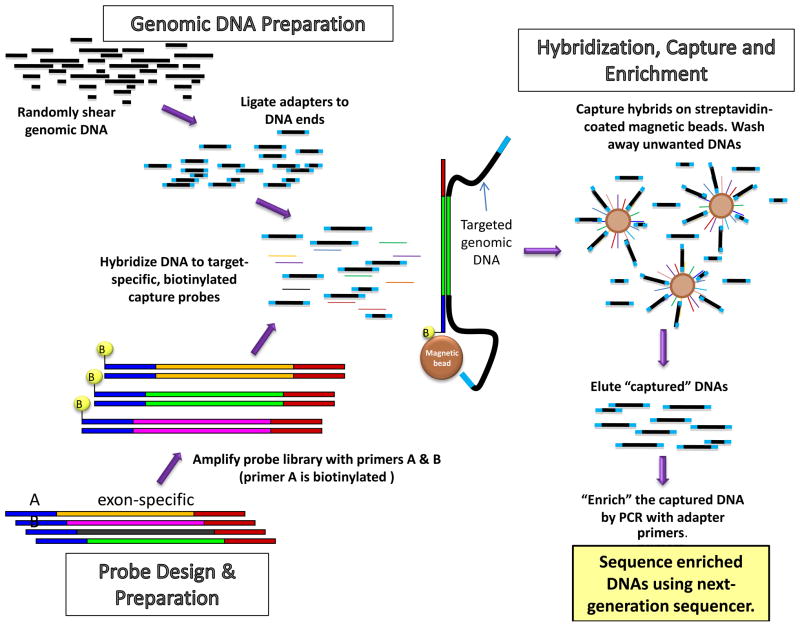
The approach to molecular genetic studies of complex phenotypes evolved considerably during the recent years. The candidate gene approach, which is restricted to an analysis of a few single-nucleotide polymorphisms (SNPs) in a modest number of cases and controls, has been supplanted by the unbiased approach of genome-wide association studies (GWAS), wherein a large number of tagger SNPs are typed in many individuals. GWAS, which are designed on the common disease-common variant hypothesis (CD-CV), identified several SNPs and loci for complex phenotypes. However, the alleles identified through GWAS are typically not causative but rather in linkage disequilibrium (LD) with the true causal variants. The common alleles, which may not capture the uncommon and rare variants, account only for a fraction of heritability of the complex traits. Hence, the focus is being shifted to rare variants-common disease (RV-CD) hypothesis, surmising that rare variants exert large effect sizes on the phenotype. In conjunctional with this conceptual shift, technologic advances in DNA sequencing techniques have dramatically enhanced whole genome or whole exome sequencing capacity. The sequencing approach affords identification of not only the rare but also the common variants. The approach-whether used in complementation with GWAS or as a stand-alone approach-could define the genetic architecture of the complex phenotypes. Robust phenotyping and large-scale sequencing studies are essential to extract the information content of the vast number of DNA sequence variants (DSVs) in the genome. To garner meaningful clinical information and link the genotype to a phenotype, the identification and characterization of a large number of causal fields beyond the information content of DNA sequence variants would be necessary. This review provides an update on the current progress and limitations in identifying DSVs that are associated with phenotypic effects.
Published by Mosby, Inc.
Conflict of interest statement
Figures

Comment in
-
High points and hurdles in the translation of genomics.Transl Res. 2012 Feb;159(2):61-3. doi: 10.1016/j.trsl.2011.10.013. Epub 2011 Nov 21. Transl Res. 2012. PMID: 22243790 No abstract available.
References
-
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. The sequence of the human genome. Science. 2001;291(5507):1304–51. - PubMed
-
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. - PubMed
-
- Lander ES. Initial impact of the sequencing of the human genome. Nature. 2011 Feb 10;470(7333):187–97. - PubMed
-
- Blaxter M. Revealing the dark matter of the genome. Science. 2010 Dec 24;330(6012):1758–9. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
