

Review
doi: 10.1016/j.tree.2011.11.010.
Epub 2012 Jan 11.
Sequencing our way towards understanding global eukaryotic biodiversity
Affiliations
- PMID: 22244672
- PMCID: PMC3311718
- DOI: 10.1016/j.tree.2011.11.010
Item in Clipboard
Review
Sequencing our way towards understanding global eukaryotic biodiversity
Trends Ecol Evol.
2012 Apr.
Abstract
Microscopic eukaryotes are abundant, diverse and fill critical ecological roles across every ecosystem on Earth, yet there is a well-recognized gap in understanding of their global biodiversity. Fundamental advances in DNA sequencing and bioinformatics now allow accurate en masse biodiversity assessments of microscopic eukaryotes from environmental samples. Despite a promising outlook, the field of eukaryotic marker gene surveys faces significant challenges: how to generate data that are most useful to the community, especially in the face of evolving sequencing technologies and bioinformatics pipelines, and how to incorporate an expanding number of target genes.
Copyright © 2011 Elsevier Ltd. All rights reserved.
Figures
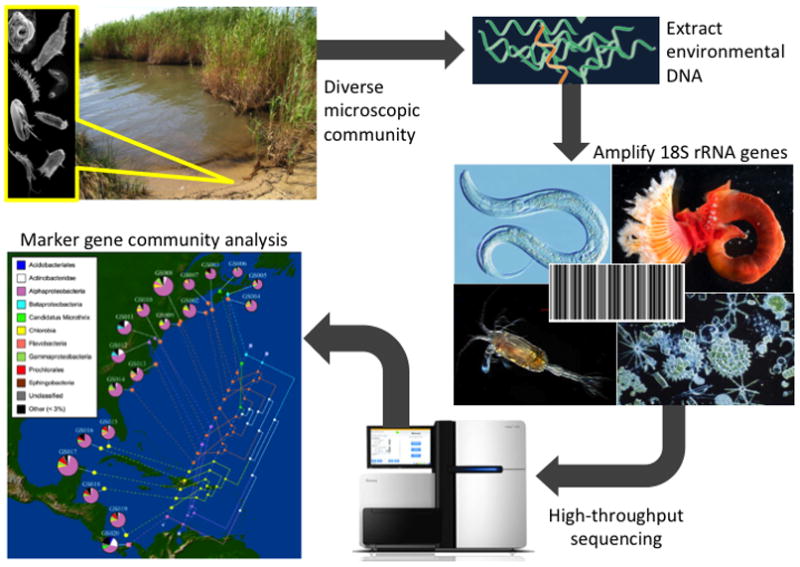
Typical standardized workflow (from environment to sequences) for high-throughput marker gene studies. Soils and sediments are typically frozen upon collection (−80°C to preserve RNA) and brought back to the lab for bulk extraction of environmental DNA. Marker genes (e.g. rRNA) are amplified from genomic extracts using barcoded, conserved primer pairs. Following high-throughput sequencing (typically conducted on 454 or Illumina platforms), datasets are processed and clustered into Operational Taxonomic Units (OTUs) under a range of pairwise identity cutoffs. OTUs are subsequently used to conduct alpha and beta diversity analyses, summarize community taxonomy, and interpret assemblages in a phylogenetic context. (Depiction of community analysis modified from Parks et al. [56])
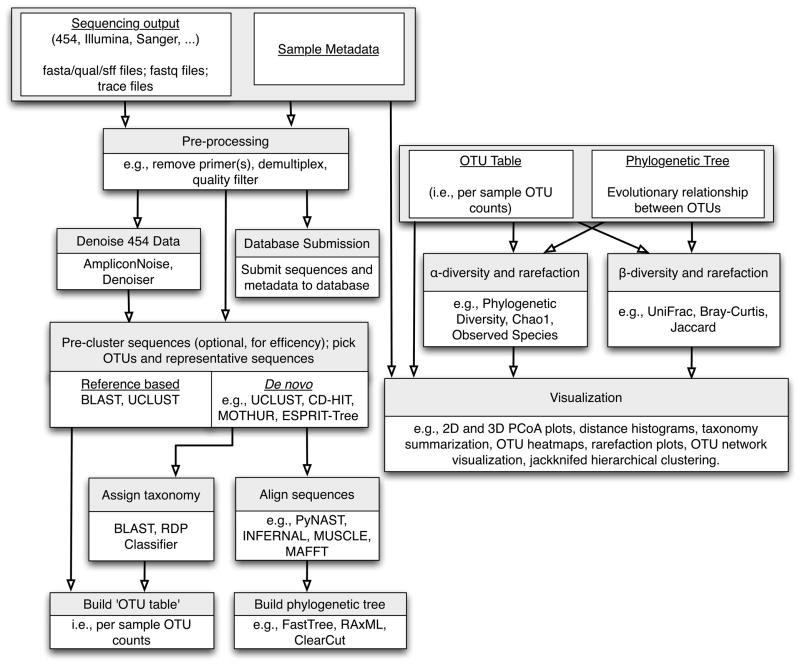
High-throughput studies follow a common workflow that begins with raw sequence data and sample metadata (primer barcodes and environmental data). Raw data is filtered and processed, with the option of denoising (a step currently applicable only to 454 data) before Operational Taxonomic Units (OTUs) are picked through reference-based or de novo approaches. OTU picking can include pre-clustering steps such as Single Linkage Preclustering (SLP, [94]), prefix-suffix filtering or collapsing of identical sequences to reduce compute time (all methods available within the QIIME pipeline [51]); the recommended and default OTU picking workflow in QIIME currently involves sorting sequences by abundance, collapsing identical reads, picking OTUs de novo with uclust, and subsequently inflating the ‘identical reads’ to recapture abundance information about the initial sequences). Taxonomy is next assigned to OTU reference sequences, followed by construction of an OTU abundance matrix and a phylogenetic tree; when working with a closed reference-based OTU picking protocol it is not necessary to make taxonomic assignments or build a phylogenetic tree as these can be obtained directly from the reference data set. These outputs can be subsequently utilized for ecological diversity analyses and visualization approaches.
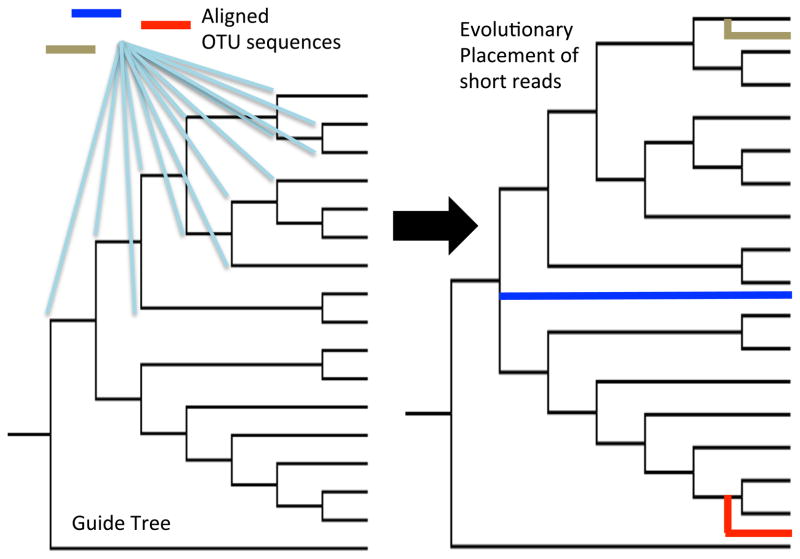
OTU reference sequences can be placed into an evolutionary context using tools such as pplacer [39] or the Evolutionary Placement Algorithm (EPA [40]), which place short reads into a guide tree framework constructed from full-length reference sequences. For each pre-aligned OTU or sequencing read, likelihood scores are calculated for all possible positions in the tree, and the sequence is subsequently inserted at the node exhibiting the best score.
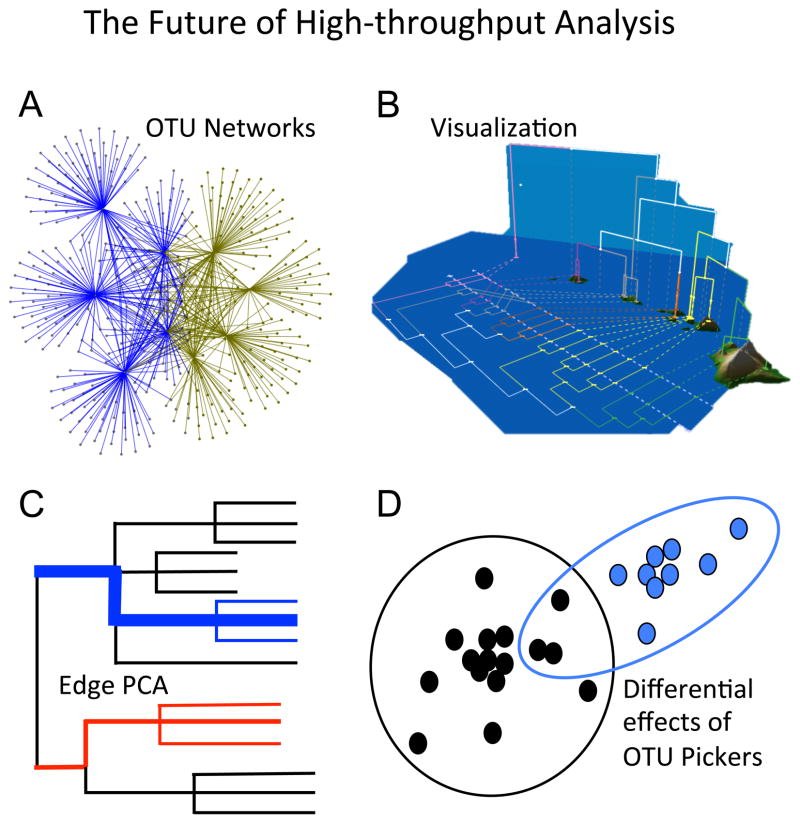
High-throughput biodiversity research is an active and rapidly evolving field. Future analytical tools will expand towards a number of exciting, emerging research areas including (A) OTU network analysis, (B) visualization as an exploratory tool (modified from Shapiro et al. [95]), (C) Edge Principal Component Analysis to defining biological lineages that define community assemblages (after Matsen & Evans), and (D) Quantifying the impact of different OTU picking strategies on cluster formation.
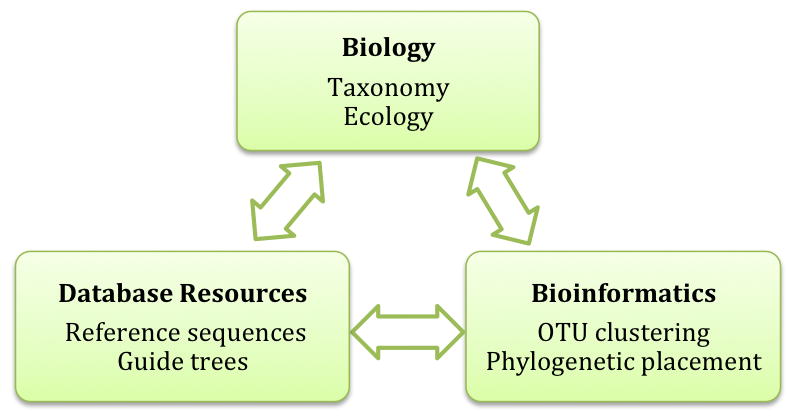
For future success, biodiversity research must adhere to a trifecta of biology, bioinformatics and database resources; none of these foci can exist in isolation, and each area must serve to inform the others. Biological questions drive high-throughput studies, and so computational pipelines and cyberinfrastructure need to functionally inform our knowledge of ecosystem processes. Likewise, computational resources must be complementary, whereby bioinformatic outputs are effectively databased, and evolving database resources produce continuing refinements in analytical pipelines. Seamless integration between these sectors will be crucial for enabling comparative metadata analyses and untangling complex ecological patterns – for example, mining published datasets for co-occurring species, or linking specific OTUs with environmental parameters (pH, salinity, temperature, etc.).
Comment in
-
Transcriptomics and microbial eukaryote diversity: a way forward.Trends Ecol Evol. 2012 Dec;27(12):651-2; author reply 652-3. doi: 10.1016/j.tree.2012.08.011. Epub 2012 Aug 31. Trends Ecol Evol. 2012. PMID: 22940223 No abstract available.
References
-
- Danovaro R, et al. Exponential decline of deep-sea ecosystem functioning linked to benthic biodiversity loss. Current Biology. 2008;18:1–18. - PubMed
-
- Wardle DA. The influence of biotic interactions on soil biodiversity. Ecology Letters. 2006;9:870–886. - PubMed
-
- Groisillier A, et al. Genetic diversity and habitats of two enigmatic marine alveolate lineages. Aquatic Microbial Ecology. 2006;42:277–291.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
